Агенезія мозолистого тіла
Агенезія мозолистого тіла (АМТ) — рідкісне вроджене захворювання, за якого спостерігається повна або часткова відсутність мозолистого тіла. АМТ може бути виявлене на МРТ, часто поєднується з різними вадами розвитку[1].
| Вроджена аномалія мозолистого тіла | |
|---|---|
 | |
| Спеціальність | неврологія |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | LA05.3 |
| МКХ-10 | Q04.0 |
| OMIM | 217990 |
| DiseasesDB | 29900 |
| MeSH | D061085 |
| | |
Класифікація
Якщо нормальний розвиток мозолистого тіла в нейроонтогенезі через певні причини було порушено, то наслідком цього можуть стати такі аномалії: агенезія мозолистого тіла (АМТ) — повна відсутність мозолистого тіла; гіпогенезія мозолистого тіла (часткова АМТ) — мозолисте тіло сформувалося частково, його відсутність повинна бути очевидною з народження й не бути проявом дегенеративного стану; дисгенезія мозолистого тіла — МТ має аномальну, нетипову форму; гіпоплазія МТ — МТ сформоване, однак має набагато менший об'єм через недостатню кількість комісуральних волокон, що беруть участь у його формуванні[2].
Розрізняють первинну й вторинну АМТ.
Відмінність гіпогенетичних змін структури МТ від вторинних ушкоджень дозволяє спертися на послідовність формування різних відділів МТ: наприклад, гіпотрофічне або відсутнє коліно за нормального тіла й валика завжди свідчить про вторинне пошкодження МТ[2].
Етіологія
Розрізняють такі причини виникнення аномалій розвитку мозолистого тіла: генетичні, інфекційні, судинні або токсичні.
Генетичні причини
Існує понад 60 генетичних і хромосомних аномалій, у яких патологія мозолистого тіла є неспецифічною ознакою.
Генетика АМТ в людей різноманітна й показує всю складність розвитку мозолистого тіла. Поєднання генетичних механізмів, в тому числі менделевської мутації одного гена, спорадичної мутації одного гена й складної генетики (що може бути поєднанням успадкованих і спорадичних мутацій) може впливати на етіологію АМТ. Причини 30—45% випадків АМТ можна встановити. У 10% присутні хромосомні відхилення, а в решти 20—35% — генетичні синдроми. У більшості людей з АМТ не простежується очевидної спадкової причини або генетичного синдрому, що дозволяє припустити, що АМТ може бути викликана випадковими новими (de novo) генетичними мутаціями.
Токсичні
Внутрішньоутробний вплив кокаїну, героїну, амфетамінів може призвести до агенезії мозолистого тіла[3].
Діагностика
Діагностика порушення здійснюється внутрішньоутробно або після народження. Методи діагностики: УЗД, МРТ, КТ.
Основні діагностичні критерії, що дозволяють визначити наявність АМТ:
1) радіальний патерн борозен медіальної поверхні мозку — борозни медіальної поверхні гемісфер мозку відходять від даху III шлуночка, поясна звивина не сформована;
2) паралельність тіл бічних шлуночків;
3) відстань між тілами бічних шлуночків збільшена;
4) аномальне розширення задніх рогів бічних шлуночків;
5) відсутність прозорої перегородки;
6) високе розташування III шлуночка.
Внутрішньоутробна діагностика АМТ
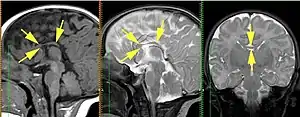
Кафедра променевої діагностики СПбДМА ім. Мечникова проводила МРТ-діагностику АМТ внутрішньоутробно. З 2004 по 2009 рік проведено 270 МРТ плодів з різною патологією, при цьому ідентифіковано 35 випадків АМТ. У 29 випадках АМТ була повною (83%), у 2 — частковою (5,7%), в 4 — атиповою (11,4%). Мозолисте тіло не визначалося в 33 випадках (94%); збільшення відстані між тілами бічних шлуночків і паралельність бічних шлуночків — 28 (80%), деформація передніх рогів бічних шлуночків — 30 (86%); розширення бічних шлуночків — 28 (80%); кольпоцефалія (аномальне розширення потиличних рогів мозку) — 27 (77%); деформація борозен медіальної поверхні гемісфер (радіальний патерн борозен) — 17 (49%). У 12 випадках (35%) АМТ була ізольованою, у 23 плодів (65%) поєднувалася з іншими аномаліями головного мозку, в 7 випадках аномалії були множинними[4].
Групи підтримки
Наразі не існує специфічного лікування вроджених аномалій мозолистого тіла. Для корекції використовуються різні методи та методики, спрямовані на допомогу у фізичному розвитку й формуванні різних навичок. Важливо проконсультуватися з різними фахівцями в галузі медицини, охорони здоров'я, освіти та соціальної роботи. До таких фахівців належать неврологи, нейропсихологи, фізіотерапевти, логопеди, педіатри, генетики, соціальні працівники, спеціальні педагоги, фахівці раннього розвитку.
Прогноз
Прогноз залежить від причини аномалії мозолистого тіла, основного захворювання та супутніх патологій. Прогноз за ізольованої форми АМТ залишається невивченим. Багато авторів вважають, що агенезія мозолистого тіла не призводить до значного порушення неврологічного розвитку. Однак величина показника специфічного ризику нині невідома[5][1].
У США організована Національна організація порушень мозолистого тіла (National Organization of Disorders of the Corpus Callosum), яка займається підтримкою осіб з порушенням розвитку мозолистого тіла, їхніх родичів. За підтримки організації проводяться щорічні тематичні конференції з участю представників наукової медичної спільноти, що займаються вивченням патологій мозолистого тіла[6].
Також організовані групи підтримки в соціальних мережах і месенджерах.
Примітки
- Timothy J. Edwards, Elliott H. Sherr, A. James Barkovich and Linda J. Richards. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes // BRAIN A JOURNAL OF NEUROLOGY. — 2014. — 01.
- А.А. Алиханов. Фенотипы нарушения структуры мозолистого тела: данные нейровизуализации (лекция) // Русский журнал детской неврологии. — 2010. — Т. 5, № выпуск 4 (23 січня).
- Dominguez, R; Aguirre Vila-Coro, A; Slopis, JM; Bohan, TP (June 1991). "Brain and ocular abnormalities in infants with in utero exposure to cocaine and other street drugs". American Journal of Diseases of Children.
- А.Д.Халиков. МРТ диагностика агенезии мозолистого тела плода // Кубанский научный медицинский вестник. — 2010. — № 6 (23 січня).
- MedUniver Агенезия мозолистого тела у плода. Диагностика и прогноз при агенезии мозолистого тела плода.
- National Organization of Disorders of the Corpus Callosum.