Молекулярний докінг
Молекулярний докінг (або молекулярне стикування) — це метод молекулярного моделювання, який дозволяє передбачити найбільш вигідну для утворення стійкого комплексу орієнтацію і положення однієї молекули по відношенню до іншої.
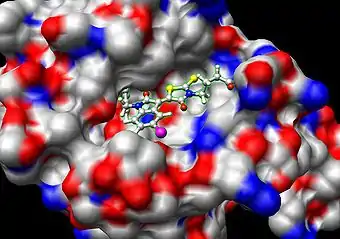
Загальний опис
Молекулярний докінг — визначення найвигіднішої орієнтації та розміщення одних молекул відносно інших.
Вихідною інформацією для докінгу слугують тривимірні структури білка (рецептора) і ліганду, конфірмаційна рухливість і взаєморозташування яких моделюється в процесі докінгу. Результатом моделювання є конформація ліганду, яка найкращим чином взаємодіє з білковим сайтом зв'язування.
Здійснюється за допомогою операції, при якій одну молекулу наближають до іншої, неперервно обчислюючи енергію взаємодії між ними при різних орієнтаціях та конформаціях, поступово встановлюючи найвигіднішу взаємну орієнтацію. При обчисленнях найчастіше враховують лише кулонівські та вандерваальсові взаємодії між атомами молекул.[1]
Знання оптимальної молекулярної орієнтації може бути використане для передбачення сили асоціації або спорідненості між двома молекулами. Зв'язок між біологічно значимими молекулами, такими як білки, нуклеїнові кислоти, вуглеводи, ліпіди відіграє центральну роль у передачі сигналу. Крім того, відносна орієнтація двох взаємодіючих партнерів може вплинути на тип сигналу, (наприклад, антагонізм проти агонізму). Тому докінг може використовуватись для прогнозування сили і типу сигналу.[2] Метод лежить в основі структурного дизайну ліків.
Основні ознаки молекулярного докінгу
- У загальному випадку число молекул довільне.
- Парний докінг — стикування однієї молекули (ліганда) до іншої (мішені).
- Моделювання дає тільки кінцевий стан комплексу (нічого не відомо про траєкторії утворення комплексу).
- У процесі роботи формується велике число проміжних варіантів надмолекуляних структур (комплексів).
- Вибір варіантів здійснюється довільно.
- Як правило процедура не дає остаточного варіанта структури комплексу.
Визначення проблеми молекулярного докінгу
Вивчення взаємодії лігандів з відповідними білками (рецепторами, ферментами) є одним з ключових завдань молекулярної біології, біотехнології та медицини, оскільки від його успішного вирішення залежить подальший прогрес у таких практично важливих областях, як розробка нових ліків, отримання високопродуктивних ферментів і тощо. Комп'ютерне моделювання міжмолекулярних взаємодій і носить назву молекулярний докінг. Схематично ідея докінгу представлена на малюнку.

Основна мета докінгу — отримання оптимальних (згідно з установленими критеріями) просторових структур комплексів. Їх аналіз дозволяє виявити ділянки «взаємного розпізнання» молекул, визначити рушійні сили, які сприяють зв'язуванню. В результаті з'являється можливість цілеспрямованого впливу на характеристики зв'язування шляхом модифікації однієї або декількох взаємодіючих молекул — наприклад, за рахунок введення точкових мутацій в білок, зміни фізико — хімічних властивостей ліганда і тощо. Як правило, при виконанні розрахунків на систему накладають певні обмеження. Наприклад, часто враховують конформаційну рухливість ліганду, тобто при обчисленні і подальшої мінімізації повної енергії системи координати атомів ліганда варіюють. Навпаки, молекулу білка, як правило, вважають нерухомою, або конформаційно лабільною є лише невелика область сайту зв'язування з лігандом. Таким чином, в процесі докінгу рухливий ліганд орієнтується відносно нерухомого білка-мішені. Взаємна просторова орієнтація ліганда і білка — мішені є основним результатом докінгу. «Правильність» орієнтації оцінюється за допомогою спеціальної оціночної функції, яка корелює з експериментально визначеною вільною енергією зв'язування ліганду. Основне призначення оціночної функції докінгу — відображати вільну енергію зв'язування ліганда (ΔΔG), проте в силу цілого ряду наближень, властивих завданням докінгу, кореляція не завжди присутня. Наведена функція враховує всі попарні невалентні (Ван — дервальсові і електростатичні) взаємодії між атомами ліганду і між лігандом і білком.[3]
Сфери застосування
Комплекси таких біологічно важливих молекул, як білки, нуклеїнові кислоти, вуглеводи і ліпіди відіграють ключову роль у передачі хімічного сигналу. До того ж, відносна орієнтація двох взаємодіючих молекул може впливати на тип генерованого сигналу (буде він інгібуючим або каталітичним). Тому докінг важливий для передбачення як типу, так і сили виробленого сигналу.
Зазвичай докінг використовують для вирішення наступних типових задач:
- Підбір лігандів, найбільш ефективно взаємодіючих з досліджуваним білком, шляхом послідовного перебору потенційних кандидатів з баз даних низькомолекулярних сполук.
- Пошук в просторовій структурі досліджуваного білка сайту зв'язування для певного ліганда.
- Оптимізація активного сайту білка — мішені шляхом введення в нього точкових мутацій, що підвищують ефективність взаємодії з певним лігандом або класом лігандів.
У першому з перелічених варіантів молекулярний докінг використовують у процесі створення нових лікарських препаратів. У варіантах 2 і 3 комп'ютерне моделювання дозволяє оптимізувати властивості білка — мішені таким чином, щоб домогтися бажаної зміни параметрів зв'язування його з потрібними лігандами — наприклад, підвищити їх афінність і / або специфічність, блокувати зв'язування і т. д.
Підходи до моделювання докінгу
Існують два підходи при моделюванні докінгу. Один підхід використовує техніку відповідності, яка описує білок і ліганд як додаткові поверхні.[4][5] Другий підхід моделює фактичний процес докінгу, в якому обчислюються попарні енергії взаємодії.[6] У обох підходах є істотні переваги, а також деякі обмеження.
Взаємозалежність форми
Геометрична відповідність (методи взаємозалежності форми) описується для білка і ліганда як ряд особливостей, які дозволяють їх докінгувати.[7] Ці особливості можуть включати як саму молекулярну поверхню, так і опис додаткових особливостей поверхні. У цьому випадку молекулярна поверхня рецептора описується з точки зору її доступності площі поверхні для розчинника, а молекулярна поверхня ліганда описується з точки зору її відповідності опису поверхні рецептора. Взаємозалежність між двома поверхнями складає опис відповідності форми, яка може допомогти виявленню додаткового положення докінга молекули-мішені і молекул ліганда.[8][9][10] В іншому підході потрібно описати гідрофобні особливості білка, використовуючи повороти в атомах головного ланцюга.
Симуляція
У цьому підході білок і ліганд відділені деякими фізичним відстаннями, і ліганд знаходить потрібне положення на активному сайті білка після певного числа «кроків». Кроки включають перетворення твердого тіла, такі як переміщення і обертання, а також внутрішні зміни структури ліганда включаючи кутові обертання. Кожен з цих кроків у просторі змінює повну енергійну оцінку системи, і отже вона обчислюється після кожного руху. Очевидна перевага цього методу полягає в тому, що це дозволяє досліджувати гнучкість ліганда під час моделювання, тоді як методи взаємозалежності форми повинні використовувати деякі інші методи, щоб дізнаватися про гнучкість ліганда. Інша перевага полягає в тому, що процес фізично ближче до того, що відбувається насправді, коли білок і ліганд наближаються до один одному після молекулярного розпізнавання. Незручність цієї техніки — те, що вона займає час, щоб оцінити оптимальну позу закріплення, так як необхідно досліджувати досить великий енергетичний пейзаж.
Програми для молекулярного докінгу
Існує багато програм для теоретичного докінгу білків. Більшість з них працює за наступним принципом: один білок фіксується в просторі, а другий повертається навколо нього різноманітними способами. При цьому, для кожної конфігурації поворотів проводяться оціночні розрахунки за оціночними функціями. Оціночна функція заснована на поверхневій комплементарності, електростатичних взаємодіях, Ван-дер-Ваальсовском відштовхуванні і так далі. Проблема при цьому пошуку в тому, що обчислення по всьому конфігураційному простору вимагають багато часу на обчислення, рідко приводячи до єдиного рішення.[11] Знання про орієнтацію можуть бути використані для передбачення міцності комплексу або спорідненості зв'язків між двома молекулами за допомогою використання окремих обчислень.
Деякі програми докінгу:
DOCK (http://dock.compbio.ucsf.edu)
- Використовується алгоритм відповідності молекулярної форми, до версії 3.5 — жорсткий докінг, починаючи з версії 4.0 — ліганд гнучкий, адаптована для пошуку лігандів в базах даних.
GOLD (http://www.ccdc.cam.ac.uk/products/life_sciences/gold/)
- Використовується генетичний алгоритм, ліганд гнучкий.
FLEXX (http://www.biosolveit.de/FlexX/)
- Ліганд гнучкий, є можливість обліку рухливості бічних радикалів амінокислотних залишків.
FRED (http://www.eyesopen.com/products/applications/fred.html)
- Жорсткий докінг, але є можливість генерації, відбору та використання конформерів. Дуже швидкий.
AUTODOCK (http://autodock.scripps.edu)
- Використовується генетичний алгоритм, ліганд гнучкий.
DOCKINGSHOP (https://web.archive.org/web/20080720140355/http://vis.lbl.gov/~scrivelli/Public/silvia_page/DockingShop.html)
Посилання
- Глосарій термінів з хімії // Й.Опейда, О.Швайка. Ін-т фізико-органічної хімії та вуглехімії ім. Л. М. Литвиненка НАН України, Донецький національний університет — Донецьк: «Вебер», 2008. — 758 с. — ISBN 978-966-335-206-0
- Wang Q, Pang YP (2007). Preference of small molecules for local minimum conformations when binding to proteins. У Romesberg, Floyd. PLoS ONE 2 (9): e820. PMC 1959118. PMID 17786192. doi:10.1371/journal.pone.0000820.
- Taylor RD, Jewsbury PJ, Essex JW (October 2003). FDS: flexible ligand and receptor docking with a continuum solvent model and soft-core energy function. J Comput Chem 24 (13): 1637–56. PMID 12926007. doi:10.1002/jcc.10295.
- Meng EC, Shoichet BK, Kuntz ID (2004). Automated docking with grid-based energy evaluation. Journal of Computational Chemistry 13 (4): 505–524. doi:10.1002/jcc.540130412.
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry 19 (14): 1639–1662. doi:10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B.
- Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks CL (2004). Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. Journal of Computational Chemistry 25 (2): 265–84. PMID 14648625. doi:10.1002/jcc.10378.
- Shoichet BK, Kuntz ID, Bodian DL (2004). Molecular docking using shape descriptors. Journal of Computational Chemistry 13 (3): 380–397. doi:10.1002/jcc.540130311.
- Cai W, Shao X, Maigret B (January 2002). Protein-ligand recognition using spherical harmonic molecular surfaces: towards a fast and efficient filter for large virtual throughput screening. J. Mol. Graph. Model. 20 (4): 313–28. PMID 11858640. doi:10.1016/S1093-3263(01)00134-6.
- Morris RJ, Najmanovich RJ, Kahraman A, Thornton JM (May 2005). Real spherical harmonic expansion coefficients as 3D shape descriptors for protein binding pocket and ligand comparisons. Bioinformatics 21 (10): 2347–55. PMID 15728116. doi:10.1093/bioinformatics/bti337.
- Kahraman A, Morris RJ, Laskowski RA, Thornton JM (April 2007). Shape variation in protein binding pockets and their ligands. J. Mol. Biol. 368 (1): 283–301. PMID 17337005. doi:10.1016/j.jmb.2007.01.086.
- Cyril Dominguez, Rolf Boelens, and Alexandre M. J. J. Bonvin (2003). HADDOCK: A Protein-Protein Docking Approach Based on Biochemical or Biophysical Information. J. AM. CHEM. SOC. 125: 1731–1737.