Нирковий тубулярний ацидоз
Ниркови́й тубулярний ацидоз - симптомокомплекс, який характеризується поліурією. рахітоподібними змінами скелету, порушенням кислотно-лужного балансу крові, відкладенням солей кальцію у печінковій паренхімі (нефрокальциноз) і утворенням в порожнині або паренхімі нирок каменів (нефролітіаз), що пов`язано з дефектами канальцевої системи нирок та нездатністю регулювати кислотно-лужну рівновагу сечі[1] . Коли кров фільтрується нирками, фільтрат проходить через канальці нефрона, що забезпечує обмін солей, кислотних еквівалентів та інших розчинених речовин , після чого він стікає в сечовий міхур у вигляді сечі. Метаболічний ацидоз, до якого призводить НТА (нирковийний тубулярний ацидоз), може бути викликаний недостатньою реабсорбцією бікарбонат іонів з фільтрату в початковій частині ниркового канальця (проксимальний каналець) або недостатньою секрецією кислоти (іонів водню) в кінцевій частині нефрона (дистальний каналець). Метаболічний ацидоз характерний для осіб з нирковою недостатністю, в той час як термін НТА використовується для людей зі зниженою здатністю нирок створювати кислу реакцію сечі, але здатних добре виконувати інші функції. Існує кілька різних типів НТА, кожен з яких має різні симптоми і причини.
Під словом ацидоз мається на увазі тенденція до зниження рН крові при НТА. Стан, за якого pH крові нижче норми (7.35) називається ацидемія. Метаболічний ацидоз, викликаний НТА належить до нормального ацидозу аніонного розриву.
| Тип | Тип 1 | Тип 2 | Тип 4 |
|---|---|---|---|
| Розташування | Збірні Канальці | Проксимальні канальці | Наднирники |
| Ацидемія | Наявна (важка) | Наявна | М'яка, якщо присутня |
| Калій | Гіпокаліємія | Гіпокаліємія | Гіперкаліємія |
| Патофізіологія | Нездатність вставних клітин типу альфа секретувати Н+ і реабсорбувати К+ | Нездатність клітин проксимальних канальців реабсорбувати НСО.3− | Дефіцит альдостерону, або резистентність до його впливу, (гіпоальдостеронізм або псевдогіпоальдостеронізм) |
Тип 1-Дистальний НТА
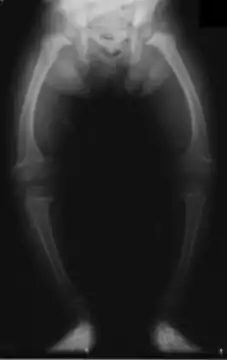
Дистальний НТА (дНТА) є класичною формою НТА, яка була описана першою. Дистальний НТА характеризується втратою здатності вставних клітин типу альфа збірних трубочок мозкової речовини дистальних відділів нефрону секретувати Н+ у просвіт нефрону. Це порушення кислотної секреції пов'язано з рядом причин і призводить до утворення сечі з рН менше за 5.3. Так як ниркова екскреції є основним засобом для усунення Н+ з організму, наслідком даного процесу є тенденція до ацидемії. Це зумовлює клінічні риси дНTA;[1] Н+/К+ антипорт на апікальній мембрані вставних клітин не функціонує, в результаті чого протони утримуються в крові, а калій екскретується. Так як кальцій фосфатні камені відкладаються при більш високих значеннях pH (лужних) дНТА сприяє їхньому формуванню в паренхімі нирок; цього не відбувається в інших видах НТА. До характерних симптомів належать:
- Нормальний метаболічний ацидоз аніонного розриву/ацидемії
- Гіпокаліємія, Гіпокальціємія, Гіперхлоремія
- Сечокам'яна хвороба (пов'язана з лужною реакцією сечі, гіперкальціурією, і низьким вмістом цитрату в сечі).[2]
- Нефрокальциноз (відкладення кальцію в паренхімі нирок)
- Демінералізація кісток (викликає рахіт у дітей і остеомаляцію у дорослих)
- Синдром Шегрена
Тип 2-Проксимальний НТА
Проксимальний НТА (пНТА) спричинений втратою здатності проксимальних тубулярних клітин реабсорбувати відфільтровані бікарбонат іони з сечі, що призводить до їхньої втрати з сечею і, як наслідок, ацидемії. Дистальні вставні клітини функціонують нормально, тому ацидемія протікає в менш важкій формі, ніж при дНТА, і сеча може закислитись до рН менше, ніж 5.3. пНТА також має декілька причин і іноді може бути присутній як одиничний дефект, але, як правило, він асоційований з більш генералізованими дисфункціями проксимальних тубулярних клітин, що має назву синдром Фанконі, за якого спостерігаються також фосфатурія, глюкозурія, аміноацидурія, урікозурія і тубулярна протеїнурія.
Характерними симптомами при синдромі Фалконі є демінералізація кісток (остеомаляція або рахіт) у зв`язку з втратою фосфатів.
Тип 3 НТА-Комбінований проксимальний і дистальний НТА
У деяких пацієнтів НTA має особливості як пНТА, так і дНTA. Ця рідкісна форма спостерігалася в 1960-ті та1970-ті роки як перехідне явище між немовлятами і дітьми з дНТА (можливо, в зв'язку з деякими екзогенними факторами, наприклад, надмірне споживання солі) і більше не спостерігається. Ця форма НTA також згадується як ювенільний НTA.
Кмбінований дНTA і пНTA може спостерігатись в результаті спадкової недостатності карбоангідрази ІІ. Мутація в гені, що кодує цей ензим призводить до виникнення аутосомного рецесивного синдрому остеопетрозу, ниркового тубулярного ацидозу, кальцифікації кінцевого мозку і розумової відсталості.[3][4][5] Цей дефект дуже рідкісний, тим не менш, були відмічені випадки по всьому світі, з яких близько 70% припадає на Магриб, регіон Північної Африки, що може буи пов`язано з високим рівнем кровної спорідненості.[6] Проблеми з нирками лікуються так, як зазначено вище. Для остеопетрозу і церебральної кальцифікації лікування не існує.
Тип 3 рідко розглядається.[7] Більшість класифікацій НТА зводяться до типів 1, 2 і 4.
Тип 4 НТА
Тип 4 насправді не є тубулярним розладом взагалі і не має клінічних синдромів, характерних для інших типів НТА, вказаних вище. Він був включений в класифікацію ниркового тубулярного ацидозу у зв`язку з тим, що він асоційований з м`яким (нормальний аніонний розрив) метаболічним ацидозом, викликаним фізіологічним зменшенням екскреції амонію в проксимальному канальці (знижений амоніагенез), що є вторинною ознакою при гіпоальдостеронізмі і призводить до зменшення буферної ємності сечі. Головною рисою даного розладу є гіперкаліємія, при якій вимірювана кислотність сечі знаходиться в межах норми, тому його часто називають гіперкаліємічним НТА або тубулярною гіперкаліємією.[7]
Історія
Льюїс постулював, що персонаж Діккенс, Тіні Тім у "Різдвяній пісні в прозі", страждав від ниркового тубулярного ацидозу.[8]
Дослідники, що опублікували PLOS One у 2009 припустили, що Карл II Зачарований міг страждати від ниркового тубулярного ацидозу в комплексі з комбінованою недостатністю гормонів гіпофізу.[9]
Див. також
Література
- Laing CM, Toye AM, Capasso G, Unwin RJ (2005). Renal tubular acidosis: developments in our understanding of the molecular basis. Int. J. Biochem. Cell Biol. 37 (6). с. 1151–61. PMID 15778079. doi:10.1016/j.biocel.2005.01.002.
- Buckalew VM Jr (1989). Nephrolithiasis in renal tubular acidosis. The Journal of Urology 141 (3 (part 2)). с. 731–737. PMID 2645431.
- Carbonic anhydrase II deficiency in 12 families with the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. N. Engl. J. Med. 313 (3). July 1985. с. 139–45. PMID 3925334. doi:10.1056/NEJM198507183130302. Проігноровано невідомий параметр
|vauthors=(довідка) - Shah GN, Bonapace G, Hu PY, Strisciuglio P, Sly WS (September 2004). Carbonic anhydrase II deficiency syndrome (osteopetrosis with renal tubular acidosis and brain calcification): novel mutations in CA2 identified by direct sequencing expand the opportunity for genotype-phenotype correlation. Hum. Mutat. 24 (3). с. 272. PMID 15300855. doi:10.1002/humu.9266.
- Molecular mechanism of kNBC1-carbonic anhydrase II interaction in proximal tubule cells. J. Physiol. (Lond.) 559 (Pt 1). August 2004. с. 55–65. PMC 1665076. PMID 15218065. doi:10.1113/jphysiol.2004.065110. Проігноровано невідомий параметр
|vauthors=(довідка) - Fathallah DM, Bejaoui M, Lepaslier D, Chater K, Sly WS, Dellagi K (1997). Carbonic anhydrase II (CA II) deficiency in Maghrebian patients: evidence for founder effect and genomic recombination at the CA II locus (PDF). Hum. Genet. 99 (5). с. 634–7. PMID 9150731. doi:10.1007/s004390050419. Архів оригіналу за 26 січня 2020. Процитовано 28 травня 2016.
- Hyporeninemic Hypoaldosteronism at eMedicine
- Lewis, Donald W. (1992). What Was Wrong with Tiny Tim?. Archives of Pediatrics & Adolescent Medicine 146 (12). с. 1403–7. PMID 1340779. doi:10.1001/archpedi.1992.02160240013002.
- Alvarez, Gonzalo; Ceballos, Francisco C.; Quinteiro, Celsa; Bauchet, Marc (15 квітня 2009). The Role of Inbreeding in the Extinction of a European Royal Dynasty. PLoS ONE 4 (4). с. e5174. PMC 2664480. PMID 19367331. doi:10.1371/journal.pone.0005174.