Множинна ендокринна неоплазія тип 2Б
Множинна ендокринна неоплазія тип 2Б (МЕН-2Б) — це генетнично обумовлене захворювання, що призводить до виникнення пухлин з локалізацією в ендокринних залозах: медулярного раку щитоподібної залози, феохромоцитоми та таких неендокринних проявів як слизові невроми, очні аномалії, ганлійнейроматоз. МЕН 2Б — це найбільш гостро протікаюче захворювання серед інших типів МЕН.[1]
Одним з перщих, МЕН-2Б описав німецький офтальмолог Августом Вагенманном, який в 1922 році опублікував спостереження про множинні невроми.[2]
| Множинна ендокринна неоплазія тип 2Б (МЕН-2Б) | |
|---|---|
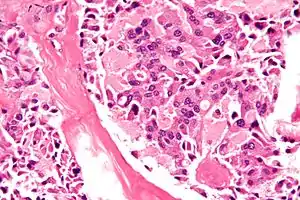 Знімок медулярного раку щитоподібної залози – однієї з складових МЕН2Б. Фарбування Г-Е. Знімок медулярного раку щитоподібної залози – однієї з складових МЕН2Б. Фарбування Г-Е. | |
| Спеціальність | ендокринологія |
| Класифікація та зовнішні ресурси | |
| OMIM | 162300 |
| DiseasesDB | 22784 |
| MeSH | D018814 |
Загальна характеристика
Класично, МЕН-2Б проявляється в віці до 10 років. При цьому доброякісні пухлини підслизової основи рота або очей діагностуються в ранньому дитячому віці, тоді як пухлини ендокринних залоз утворюються в пубертатному віці. Медулярний рак щитоподібної залози діагностується в усіх випадках, тоді як феохромоцитома — до 50%. Слід зазначити, що медулярний рак характеризується високим показником інвазивності, що може призводити до летальних випадків ще до розвитку інших новоутворень ендокринних органів. Рівень захворюваності на МЕН-2Б становить 1 на 40000 населення.[3]
Етіологія
МЕН-2Б — це спадкове захворювання, що передається за автосомно-домінантним типом, що означає наявність хімерного гена у одного з батьків, внаслідок чого МЕН-2Б можуть хворіти не тільки діти, але і сіблінги. Проте, до 50% МЕН-2Б характеризуються спонтанним мутаціями. 95% батьків з МЕН-2Б мають мутацію в гені RET.[4] Ця мутація виникає внаслідок заміни метіоніну на треонін в внутрішньоклітинному домені тирозин-кінази.[5]
Також, до 50% випадків МЕН-2Б можуть виникати як спорадичні випадки, тобто внаслідок мутацій de novo , що виникли внаслідок генетичної помилки в ДНК сперматозоїда або яйцеклітини ще до запліднення. Як правило, пацієнти з de novo мутацією народились у літніх батьків, зокрема чоловіків, таким чином вік батьків може бути фактором ризику щодо розвитку МЕН-2Б.[1]
Посилання
- Carlson KM, Bracamontes J, Jackson CE, et al. (December 1994). Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am. J. Hum. Genet. 55 (6): 1076–82. PMC 1918453. PMID 7977365.
- Wagenmann A. (1922). Multiple neurome des Auges und der Zunge. Ber Dtsch Opthalmol Ges: 282–5. Проігноровано невідомий параметр
|vol=(довідка) - Martino Ruggieri (2005). Neurocutaneous Disorders : The Phakomatoses. Berlin: Springer. ISBN 3-211-21396-1. — Chapter: Multiple Endocrine Neoplasia Type 2B by Electron Kebebew, Jessica E. Gosnell and Emily Reiff. Pages 695–701.
- Sperling, Mark A. (2008). Pediatric Endocrinology (вид. 3). Elsevier Health Sciences. с. 246–7. ISBN 1-4160-4090-0.
- Morrison PJ, Nevin NC (September 1996). Multiple endocrine neoplasia type 2B (mucosal neuroma syndrome, Wagenmann-Froboese syndrome). J. Med. Genet. 33 (9): 779–82. PMC 1050735. PMID 8880581. doi:10.1136/jmg.33.9.779.