Реакція Хорнера — Водсворта — Еммонса
Реа́кція Хо́рнера — Во́дсворта — Е́ммонса (також олефінування Хорнера — Водсворта — Еммонса) — реакція отримання олефінів з альдегідів (або кетонів) та фосфонатних карбаніонів[1][2]. Під час реакції утворюються переважно транс-(або (Е)-)алкени.
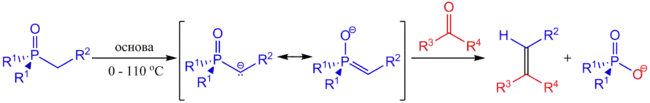
Реакція синтезу олефінів з використанням ілідів Фосфору була вперше запропонована в 1953 році Георгом Віттігом і Гейслером. У 1958 році німецький хімік Леопольд Хорнер опублікував удосконалену реакцію Віттіга з використанням карбаніонів алкілдифенілфосфіноксидів для отримання алкенів з альдегідів та кетонів. Цю модифікацію реакції Віттіга назвали реакцією Хорнера—Віттіга. Пізніше, на початку 1960-х років, Вільям Водсворт та Вільям Еммонс удосконалили умови реакції використовуючи фосфонатні карбаніони. Тому реакцію назвають реакцією Хорнера—Водсворта—Еммонса або ж олефінуванням Хорнера—Водсворта—Еммонса[3].
Механізм реакції
Реакція Хорнера—Водсворта—Еммонса починається з депротонування фосфонату з утворенням фосфонатного карбаніону. Лімітуючою стадією є нуклеофільне приєднання карбаніону до альдегіду (або кетону) з утворенням проміжних інтермедіатів. Останнім етапом є елімінування інтермедіату з утворенням (Е)-алкену та (Z)-алкену. Співвідношення (Е)- та (Z)-ізомерів алкену залежить від стереохімічного виходу початкової стадії приєднання карбаніону та від здатності інтермедіатів еквілібрувати. У результаті реакції Хорнера—Водсворта—Еммонса переважно утворюються (Е)-алкени. Загалом, чим більше урівноважування інтермедіатів, тим вища селективність до утворення (Е)-алкенів.
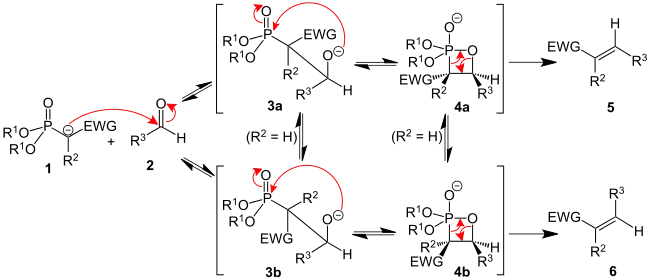
Електроноакцепторна група в альфа-положенні до фосфонату забезпечує заключне елімінування. За відсутності електроноакцепторної групи продуктом реакції є α-гідроксифосфонати[4]. Однак такі α-гідроксифосфонати можуть бути перетворені на алкени з допомогою реакції з диізопропілкарбодиімідом[5].
Переваги реакції Хорнера—Водсворта—Еммонса
Реакція Хорнера—Водсворта—Еммонса має низку переваг порівняно з реакцією Віттіга:
- процедура отримання початкових алкілфосфонатів є легшою (зазвичай використовують реакцію Арбузова) та дешевшою, ніж отримання фосфонієвих солей;
- фосфонатні стабілізовані карбаніони є більш нуклеофільними (і більш основними), ніж відповідні трифенілфосфонієві іліди у реакції Віттіга, тому вони легко реагують практично з усіма альдегідами і кетонами при м'якших умовах;
- заміщені кетони, які не є реакційноздатними у реакції Віттіга, досить легко вступають у реакцію олефінування Хорнера-Водсворта-Еммонса;
- α-кетон фосфонатного аніону може бути й далі функціоналізований різними електрофілами (наприклад, алкілгалогенідами) перед олефінуванням, тоді як фосфонієві іліди не піддаються такому м'якому алкілюванню;
- супутні продукти реакції Хорнера—Водсворта—Еммонса — диалкілфосфати, які легко відмиваються водою, тому значно простіше відділити їх отриманих алкенів, ніж відділити нерозчинний у воді трифенілфосфіноксид[6].
Основними характеристиками реакції олефінування Хорнера—Водсворта—Еммонса є:
- висока (Е)-селективність для дизаміщених алкенів при значно м'якших умовах ніж у реакції Віттіга;
- (Е)-селективність збільшується при збільшення розміру алкільних замісників R1 та R2 (найкращим є використання сполук з R — ізопропіл);
- стереоселективність дуже сильно залежить від субстрату, але може бути обернена й утворювати переважно (Z)-алкени при використання сполук з малими за розміром алкільними замісниками R1 та R2 (наприклад, метильними) та при використання сильно дисоційованої основи (трет-бутоксид Калію)[3].
Модифікації реакції Хорнера—Водсворта—Еммонса
Модифікація Стілла—Дженаррі
Стілл та Дженнарі розробили умови, в результаті яких продуктами реакції є (Z)-алкени з високим стереохімічним виходом[7]. Цього вдалося досягти за рахунок використання фосфонатів з електроноакцепторними групами (трифлуороетоксидними групами)[8] в умовах сильної дисоціації (Калію гексаметилдисилазид та 18-краун-6 в тетрагідрофурані). Андо також зауважив, що використання елекронодефіцитних груп прискорює відщеплення оксафосфатанових інтермедіатів.[9]

Реакція Хорнера—Водсворта—Еммонса з використанням чутливих до основ субстратів
Оскільки багато субстратів не є стійкими до гідриду Натрію, були розроблені декілька методик з використанням м'якших основ. Масам'юн та Рауш пом'якшили умови за рахунок використання хлориду літію (або йодиду натрію) та диазабіциклоундецену[10]. Ратке запропонував використовувати галогеніди літію або магнію з триетиламіном[11]. Такі умови дозволяють уникнути епімеризації.
Асимеричні реакції олефінування Хорнера—Водсворта—Еммонса
У цьому випадку опираються на три основні принципи:
- введення С2-симетричного хірального центру замість алкоксидних груп у фосфонатах;
- введення С2-несиметричного хірального центру замість алкоксидних груп у фосфонатах, що зробить хіральним сам атом Фосфору;
- введення хірального центру в ту частину фосфонату, яка переміщається на карбонільну сполуку[12].
Модифікація Корі—Квятковскі
У модифікації Корі—Квятковські використовують бісаміди фосфонатної кислоти для стереоселективного отримання (Z)-алкенів, (Me2N)2P(O)CH2R, де R — арильний замісник[13][14].
Див. також
Примітки
- Horner, L., Hoffman, H., Wippel, H. G., Klahre, G. Chem. Ber. 1959, 92, 2499.
- Wadsworth, W. S., Jr., Emmons, W. D. J. Am. Chem. Soc. 1961, 83, 1733.
- Wadsworth, D. H., Schupp, I. O. E., Sous, E. J., Ford, J. J. A. J. Org. Chem. 1965, 30, 680.
- Corey, E. J.; Kwiatkowski, G. T. J. Am. Chem. Soc. 1966, 88, 5654.
- Reichwein, J. F.; Pagenkopf, B. L. J. Am. Chem. Soc. 2003, 125, 1821.
- Kürti L., Czakó B. Strategic Applications of Named Reactions in Organic Synthesis, Academin Press, 2005.
- Still, W. C.; Gennari, C. Tetrahedron Lett. 1983, 24, 4405.
- Patois, C.; Savignac, P.; About-Jaudet, E.; Collignon, N. Organic Syntheses, 1998, 9, 88; 1996, 73, 152.
- Ando, K. J. Org. Chem. 1997, 62, 1934.
- Blanchette, M. A.; Choy, W.; Davis, J. T.; Essenfeld, A. P.; Masamune, S.; Roush, W. R.; Sakai, T. Tetrahedron Lett. 1984, 25, 2183.
- Rathke, M. W.; Nowak, M. J. Org. Chem. 1985, 50, 2624.
- Rein, T.; Reiser, O. Acta Chem. Scand. 1996, 50, 369.
- Corey, E. J., Kwiatkowski, G. T. J. Am. Chem. Soc. 1966, 88, 5652.
- Corey, E. J., Kwiatkowski, G. T. J. Am. Chem. Soc. 1968, 90, 6816.
Джерела
- Clayden J., Greeves N., Warren S. Organic chemistry. — 2nd ed. — Oxford University Press, 2012. — P. 628. — ISBN 978-0-19-927029-3.
- Kürti, László; Czakó, Barbara (2005). Strategic Applications of Named Reactions in Organic Synthesis. Elsevier Academic Press. с. 864. ISBN 0-12-429785-4.(англ.)
Посилання
- Реакція Хорнера-Водсворта-Еммонса на сайті organic-chemistry.org (англ.)