Синдром Аперта
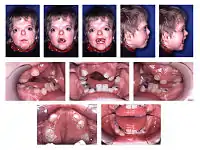
Синдро́м А́перта, або Акроцефалосиндактилія I типу[1] (англ. Apert Syndrom) — це один з типів акроцефалосиндактилії, що проявляється вадами розвитку обличчя, кісток черепу, пальців рук та ніг.[2] Частіше зустрічається у нащадків літніх людей.[3]
| Синдром Аперта | |
|---|---|
 | |
| Спеціальність | медична генетика |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | LD24.G2 |
| МКХ-10 | Q78.0 |
| OMIM | 101200 |
| DiseasesDB | 33968 |
| MedlinePlus | 001581 |
| eMedicine | ped/122 |
| MeSH | D000168 |
| SNOMED CT | 205258009 |
| | |
Історичні відомості
Перші згадки про цю хворобу відносяться до 1894 року.
У 1906 році французький педіатр Євген Аперт систематизував спостереження цієї аномалії, та описав прояви хвороби.
До 1960 року було 150 зафіксованих документально випадків цього синдрому.[1]
Етіологія
Синдром Аперта — це автосомно-домінантна патологія. Приблизно дві третини випадків захворювання пов'язані з мутацією, що змінює цитозин (Ц) на гуанін (Г) у 755-ій позиції мРНК гену FGFR2 і призводить до заміни амінокислотного залишку серину на триптофан у білку.[4] Білок FGFR2 є мембранним рецептором до фактору росту фібробластів і бере участь у формуванні кісток та загоєнні ран.
Примітки
- http://www.ibis-birthdefects.org/start/ukrainian/uacrocep.htm
- Fearon, Jeffrey A. (July 2003). Treatment of the Hands and Feet in Apert Syndrome: An Evolution in Management. Plastic and Reconstructive surgery (Dallas, Texas) 112 (1): 1–12. ISSN 0032-1052. PMID 12832871. doi:10.1097/01.PRS.0000065908.60382.17.
- http://pan-ta-pani.com/92084-hromosomni-anomali-i-litnijj-vik-batka.html
- Goriely, A.; McVean, GA; Röjmyr, M; Ingemarsson, B; Wilkie, AO (2003). Evidence for Selective Advantage of Pathogenic FGFR2 Mutations in the Male Germ Line. Science 301 (5633): 643–6. PMID 12893942. doi:10.1126/science.1085710.
Посилання
- Синдром Аперта Інформація від Seattle Children's Hospital Craniofacial Center (англ.)
- Синдром АПЕР у плода. Діагностика і прогноз при синдромі АПЕР
- Акроцефалосиндактилії
