Синдром Жильбера
Синдро́м Жильбе́ра (також — проста сімейна холемія, конституційна гіпербілірубінемія, ідіопатична некон'югована гіпербілірубінемія, негемолітична сімейна жовтяниця) — пігментний гепатоз, синдром з автосомно-рецесивним (зрідка автосомно-домінантним) типом успадкування, що характеризується підвищенням рівня непрямого білірубіну в крові (гіпербілірубінемія) внаслідок порушення внутрішньоклітинного транспорту білірубіну в печінкових клітинах (гепатоцитах) до місця його з'єднання з глюкуроновою кислотою, зменшенням рівня гіпербілірубінемії під дією фенобарбіталу. Також синдром визначають як доброякісну сімейну некон'юговану гіпербілірубінемію помірної виразності, яка не пов'язана з гемолізом еритроцитів.
| Синдром Жильбера | |
|---|---|
 | |
| Спеціальність | гепатологія і сімейна медицина |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | 5C58.01 |
| МКХ-10 | E80.4 |
| OMIM | 143500 |
| DiseasesDB | 5218 |
| MedlinePlus | 000301 |
| eMedicine | med/870 |
| MeSH | D005878 |
| SNOMED CT | 27503000 |
Історичні відомості
Це доброякісне хронічне ураження вперше описали у 1900—1901 роках французькі лікарі-терапевти Августин Ніколя Жильбер і Пьєр Леребулле.[1][2] На честь Жильбера дали назву цьому синдрому[3], однак сучасні німецькі лікарі з цим не згодні, вони вважають, що синдром вперше описав у 1939 році німецький лікар Йенс Мейленграхт[4], тому називають ураження відповідно «синдромом Мейленграхта».[3]
Епідеміологічні дані
Найчастіша форма спадкового пігментного гепатозу, яку виявляють у 1-5 % населення. Синдром поширений у європейців (2-5 %), азіатів (3 %) і африканців (36 %). Синдром проявляється зазвичай у підлітковому періоді. Часто хворі не підозрюють про свою хворобу, поки не виявиться при клінічному огляді чи при проведенні лабораторних досліджень[5].
Генетичні порушення
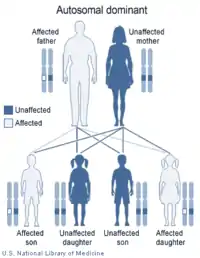
Синдром має аутосомно-домінантний тип успадкування.[6] Ген, що кодує УДФ-глюкуронозілтрансферазу-1 (UGT1A1) має промотор-TATA-Box, який містить алелі A(TA)6TAA. Синдром, як правило, асоціюється з гомозиготними A(TA)7TAA алелями.[7] Поліморфізм алелей позначають UGT1A1*28.
Патогенез
Через доброякісність перебігу синдрому в людей ґрунтовні дослідження з вивчення патогенезу проведені на тваринних моделях — болівійських білячих мавпах. У 94 % при хворобі Жильбера пошкоджуються ферменти сім'ї глікозилтрансфераз: UDP-GT1-A6 (зниження активності приблизно на 50 %) і UDP-GT1-A7 (зниження активності приблизно на 83 %). Внаслідок цього відбувається порушення захоплення білірубіну мікросомами васкулярного полюсу печінкової клітини (гепатоцита), порушення його транспортування глутатіон-S-трансферазою, яка доставляє непрямий білірубін до мікросом гепатоцитів, а також неповноцінність мікросомального ферменту урідиндифосфатглюкуронілтрансферази, за допомогою якої відбувається кон'югація непрямого білірубіну з глюкуроновою та іншими кислотами. Через усі ці порушення при синдромі Жильбера зменшується ще й активність УДФ-глюкуронілтрансферази на 70-75 %.[8][9] У жовчі виявляють переважно білірубіна моноглюкуронід, меншою мірою — диглюкуронід. Надлишковий непрямий білірубін, який на відміну від прямого білірубіну, не здатний виходити з сечею, внаслідок притаманної йому тропності потрапляє до центральної нервової системи, де спричинює гальмування роботи деяких структур.
Клінічні ознаки
Провокує загострення синдрому голодування, інтеркурентні інфекційні захворювання, тривале блювання з будь-яких причин. При невисокому рівні білірубіна в крові пацієнтів практично нічого не турбує. Під час загострення, коли рівень непрямого білірубіна відчутно підвищується, пацієнти скаржаться на немотивовану слабкість, швидку втомлюваність, нудоту, дискомфорт у ділянці печінки, гальмування когнітивних процесів, неможливість виконання інтенсивної розумової роботи тощо. Однак перебіг синдрому доброякісний, тривалості життя не зменшує. Іноді пацієнтів взагалі нічого не турбує і виявлення синдрому відбувається випадково, під час обстеження з приводу інших хвороб або при проведенні медичного профілактичного огляду.
Діагностика
Спеціальні методики визначення генетичних аномалій, стану ферментних систем печінкових клітин є складними для рутинної клінічної практики. Використовують діагностичні проби: одноденного голодування, що призводить до збільшення в крові рівню непрямого білірубіна, або з фенобарбіталом, вживання якого в дозі 0,1 г наступного дня після прийому призводить до зменшення рівню непрямого білірубіна в крові.
Лікування
У цілому перебігає доброякісно, через що призначення препаратів має бути під час загострення, підвищення рівню непрямого білірубіну. Призначають фенобарбітал, але коротким курсом, аби не спричинити розвиток барбітурової залежності.
Примітки
- A. Gilbert, M. J. Castaigne, P. Lereboullet: De l'ictère familial. Contribution à l’étude de la diathèse biliaire. Bulletin de la Société des médecins des hôpitaux de Paris, 1900, 17: 948—959. Castaigne and Lereboullet were colleagues of Gilbert.
- A. Gilbert, P. Lereboullet: La cholémie simple familiale. Semaine médicale, Paris, 1901, 21: 241—243.
- Whonamedit? — A dictionary of medical eponyms. Gilbert's syndrome
- E. Meulengracht: Icterus intermittens juvenilis (chronischer intermittierender juveniler Subikterus). Klinische Wochenschrift, Berlin, 1939, 45: 118—121.
- S.Lejniece. Гемолітичні анемії. // Внутрішня медицина. — 2009. № 5-6. с. 17-18
- Anthony S. Fauci u. a.: Harrison's Principles of Internal Medicine. 17. Auflage. McGraw-Hill, New York 2008, ISBN 0-07-146633-9, S. 1929.
- G. Monaghan u. a.: Genetic variation in bilirubin UPD-glucuronosyltransferase gene promoter and Gilbert’s syndrome. In: Lancet. 347, Nr. 9001, 1996, S. 578–581 (DOI:10.1016/S0140-6736(96)91273-8).
- Maarten Raijmakers, Peter Jansen, Eric Steegers, Wilbert Peters: Association of human liver bilirubin UDP-glucuronyltransferase activity, most commonly due to a polymorphism in the promoter region of the UGT1A1 gene. In: Journal of Hepatology. 33, Nr. 3, 2000, S. 348–351 (DOI:10.1016/S0168-8278(00)80268-8).
- Piter J. Bosma u. a.: The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. In: New England Journal of Medicine. 333, Nr. 18, 1995, S. 1171–1175 (DOI:10.1056/NEJM199511023331802).
Література
- Ш. Шерлок, Дж. Дули Заболевания печени и желчных путей (пер. с англ.) (под ред. З. Г. Апросиной, Н. А. Мухина). — 1999, М. «ГЭОТАР, Медицина». — 864 с. ISBN 5-88816-013-Х (рос.)