Полікістоз нирок
Полікістоз нирок — спадкове захворювання, яке спричинюють генетичні мутації, що характеризується утворенням численних кіст в паренхимі нирок та поступовому їх збільшенні[1]. Захворювання передається спадково, виникнення його внаслідок випадкової мутації, тобто спорадично, відбувається зрідка. Полікістоз нирок одна з найпоширеніших смертельно небезпечних генетичних хвороб, від неї страждають більше 12,5 мільйонів людей у світі[2].
| Полікістоз нирок | |
|---|---|
 | |
| Спеціальність | медична генетика |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | GB81 |
| OMIM | 173900 |
| | |
Класифікація
Полікістоз нирок успадковується по аутосомно-домінантному чи рецесивному типу. За цією ознакою він поділяється на 2 типи — аутосомний домінантний полікістоз нирок (АДПН) і аутосомний рецесивний полікістоз нирок (АРПН). Кожен з них має свої власні причини виникнення і патології.
Аутосомний домінантний полікістоз нирок — найбільш поширене спадкове кістозне захворювання нирок[3]. Воно зустрічається у 1 з 500 народжених немовлят[4]. За статистичними даними в 10 % пацієнтів, яким робили гемодіаліз у Європі і США, на пізніх стадіях ниркової недостатності, її спричинив саме — АДПН. За результатами досліджень не було виявлено кореляції між частотою захворювання на АДНП та расовою чи етнічною приналежністю.

АДПН характеризується прогресивним утворенням кіст і, як наслідок цього, збільшенням нирок. Він спричинений трьома мутаціями у генах PKD-1, PKD-2, PKD3. Ген PKD-1 розташований у 16 хромосомі й кодує білок, який включається в регуляцію клітинного циклу і внутрішньоклітинного транспорту кальцію у епітеліальних клітинах. Мутація в цьому гені слугує причиною виникнення АДНП у 85 % випадків. З гену PKD-2 експресуються білки, що входять до складу групи потенціал-залежних кальцієвих каналів. Він локалізується на 4 хромосомі. PKD3 був відкритий зовсім недавно і наразі досліджений недостатньо. АДПН починає проявлятися у віці 30-40 років.
Аутосомний рецесивний полікістоз нирок — набагато рідкісніше захворювання, зустрічається з частотою 1 до 20000 і проявляється на перших тижнях після народження. Причиною захворювання є мутація гену PKHD1, який кодує білок фіброцистин. Через розвиток хвороби рівень летальності в новонароджених сягає 30 %. При АРПН нирки зберігають свою форму, але відбувається деформація канальців — їх розширення від мозкової речовини до кори нирки.
Етіологія
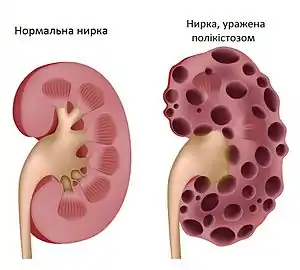
Полікістоз нирок характеризується наявністю численних кіст в обох нирках, рідше в одній (17 % від усіх випадків захворювання)[5]. Кісти утворюються в результаті підвищеної проліферації і диференціації епітелію ниркових канальців. Наслідком цього замість нормального ниркового канальця утворюється пухирець, наповнений рідиною — кіста. Це призводить до збільшення об'єму і маси нирки (вага нирки хворого може сягати 35 кг). Кісти в нирці виникають фокально, їх формують не більше ніж 2-5 % всіх нефронів, але через збільшення об'єму кіст відбувається здавлення сусідніх здорових клітин. Внаслідок цього розвивається ниркова недостатність. АДПН і АРПН відносяться до ціліопатій — група захворювань, за яких порушується нормальна робота війок на поверхні клітин. Білки поліцистин-1, поліцистин-2 і фіброцистин входять до складу таких війок. У клітинах ниркового епітелію первинні війки розміщуються зі сторони просвіту ниркових канальців і, за припущеннями, виконують сенсорну функцію — реагують на тік сечі. Через неправильне сприйняття сигналів через порушення роботи первинних війок у клітинах ниркового епітелію накопичується циклічний аденозинмонофосфат. На пошук шляхів зниження його концентрації направлені сучасні експериментальні дослідження.
Клінічні ознаки
Зазвичай ознаки хвороби виявляються не відразу. Перші прояви полікістозу нирок виникають, коли маса функціонуючої паренхіми значно зменшується. Тоді, перш за все, порушується реабсорбційна функція нирки. Наслідком цього є поліурія (сягає 3-4 л на добу), постійне відчуття спраги. За цими симптомами слідують інші: погіршення апетиту, зниження працездатності, тупий біль і почуття важкості у ділянці попереку, головний біль, гематурія. При пальпації визначається збільшена, горбиста, щільна нирка, що при натисканні болить[6]. З часом порушується виведення нирками азотовмісних сполук і виникає азотемія. Це призводить до погіршення стану хворого, з'являються нові симптоми: неприємний смак у роті, нудота. Полікістоз нирок часто супроводжується артеріальною гіпертензією, що сприяє прогресуванню хвороби і збільшує тяжкість її перебігу. На пізніх стадіях захворювання спостерігається розвиток анемії та інші ознаки ниркової недостатності.
Діагностика та лікування
Діагностика включає детальне опитування пацієнта, стосовно його скарг, наявності подібного захворювання у кровних родичів. Лабораторні дослідження включають — клінічний аналіз сечі і клінічний аналіз крові, біохімічний аналіз крові, аналіз сечі по Зимницькому, генетичний аналіз. Найчастіше використовуються такі інструментальні методи дослідження, як ультразвукове обстеження, внутрішньовенна орографія (дослідження нирок і сечових шляхів за допомогою рентгенівського просвічування з використанням контрастної речовини), комп'ютерна томографія[7]. Вони допомагають виявити виражені кістозні зміни в паренхімі нирок і «рваний» вигляд, що обумовлюється великою кількістю кіст. Лікування є симптоматичним, тобто направлене на усунення або полегшення проявів хвороби. Застосовуються знеболювальні, антигіпертензивні препарати, за наявності розвитку патогенних організмів у порожнинах кіст, паренхімі нирок, сечовивідних шляхах — приписують вживання антибіотиків. Хворому потрібно дотримуватись суворої дієти, яка обмежує вживання солі, білкової їжі, рідини. Також існує практика оперативних втручань. Її застосовують при наявності великих кіст, вираженій кровотечі чи сильному мікробному запаленні. Ці хірургічні маніпуляції полягають у видаленні вмісту кісти, її самої чи взагалі нефректомії. На пізніх стадіях хворим необхідне проведення гемодіалізу.
Примітки
- Polycystic kidney disease at Dorland's Medical Dictionary
- Wilson P.D. Polycystic kidney disease / Wilson P.D.// N.Engl.J.Med. — 2004. — Vol.350. — P.151-164.
- Torres W.E."Autosomal dominant polycystic urology" / Torres W.E., Harris P.C., Pirson Y. // Lancet. — 2007. — Vol.369(9569). — P.1287-301
- Simons M. Polycystic kidney disease: cell division with a c(l)ue? / Simons M., Walz G. // Kidney International. — 2006. — Vol.70. — P.854-864.
- Bisceglia M. Renal cystic diseases: a review / Bisceglia M. et al. // Advanced Anatomic Pathology. — 2006. — Vol.13. — P.26-56.
- (рос.)Поликистозная болезнь почек (Поликистоз почек)
- (рос.)ПОЛИКИСТОЗ ПОЧЕК — ЛЕЧЕНИЕ РАЗЛИЧНЫМИ МЕТОДАМИ Архівовано 14 травня 2014 у Wayback Machine.