Синдром Муїр-Торре
Синдром Муїр-Торре — рідкісний спадковий, синдром аутосомно-домінантного раку[1] який вважається підтипом спадкового неполіпозного колоректального раку (HNPCC). Хворі схильні до розвитку ракових уражень товстої кишки, сечостатевих шляхів та новоутворень шкіри, таких як кератоакантоми та пухлини сальних залоз. Уражені гени — MLH1, MSH2, MSH6, що беруть участь у репарації помилково спарених нуклеотидів.
| Синдром Муїр-Торре | |
|---|---|
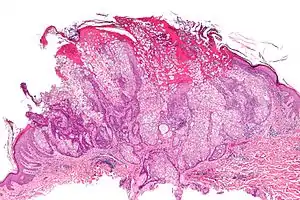 Мікрофотографія себорейної аденоми, яка може спостерігатися при синдромі Муїр-Торре. Фарбування Г-Е. Мікрофотографія себорейної аденоми, яка може спостерігатися при синдромі Муїр-Торре. Фарбування Г-Е. | |
| Інші назви | MTS |
| Спеціальність | онкологія, дерматологія і медична генетика |
| Класифікація та зовнішні ресурси | |
| OMIM | 158320 |
| DiseasesDB | 31385 |
| MeSH | D055653 |
Симптоми
Синдром Муїр-Торре характеризується двома симптомами:[2]
- Принаймні одна пухлина сальної залози (аденома, епітеліома, карцинома)
- Мінімум одна внутрішня злоякісна пухлина
Амстердамські критерії часто використовуються для діагностики синдрому Лінча та синдрому Муїр-Торре. До них належать такі ознаки:
- троє або більше родичів з HNPCC-асоційованим раком (наприклад: колоректальним раком, раком ендометрію, раком тонкої кишки, раком сечоводу, раком ниркової миски)
- два або більше послідовних покоління, уражених раком
- одна або більше особа, хвора на рак, є родичами першого ступеня інших двох, з, принаймні, одним випадком колоректального раку молодше 50 років, діагноз сімейного аденоматозного поліпозу виключений, пухлини верифіковані гістологічним дослідженням
Синдром Муїр-Торре — генетична хвороба. Мутації в MLH1 та MSH2 пов'язані з цим захворюванням. Ці гени кодують білки, що беруть участь у репарації помилково спарених нуклеотидів, їх мутації підвищують ризик розвитку пухлин.
Багато пацієнтів, які мають новоутворення сальних залоз з мутаціями в MSH2 та MLH1, насправді не мають синдрому Муїра-Торре.
Шкала ризику Майо була розроблена для поліпшення позитивного прогностичного значення імуногістохімії та для зниження частоти помилково позитивних діагнозів.[3][4] За шкалою ризику Майо бали присвоюються на основі декількох характеристик. Оцінка 2 або більше вказує високий позитивний прогноз синдрому Муїра-Торре. Оцінка 0-1 вказує на менший ризик.
- Вік виникнення першої сальної пухлини: <60 років = 1 бал, інакше 0 балів
- Загальна кількість новоутворень сальних залоз: 1 = 0 балів, >2 = 2 бали.
- Особистий онкологічний анамнез, пов'язаний з синдромом Лінча: Ні = 0 балів, Так = 1 бал
- Сімейний онкологічний анамнез, пов'язаний з синдромом Лінча: Ні = 0 балів, Так = 1 бал
Найпоширенішими внутрішніми злоякісними новоутвореннями, пов'язаними з синдромом Муїр-Торре, є: колоректальний рак (56 %), сечостатевий рак (22 %), рак тонкого кишечника (4 %) та рак молочної залози (4 %). Повідомлялося про низку інших внутрішніх злоякісних новоутворень.[5]
Етіологія
Генетичне перекриття з синдромом Туркота
Було проведено декільки досліджень на пацієнтах із синдромом Муїр-Торре та синдромом Туркота. Вважається, що вони можуть мати певне генетичне перекриття. В обох випадках були пов'язані дефекти генів MLH1 та MSH2.[6]
В одному дослідженні у пацієнта з дефектними генами репарації MSH2 та MSH6 виявлено обидва синдроми. Це перший випадок, коли у пацієнта зі змінами генотипу, що відповідають HNPCC, було належним чином діагностовано перекриття обох синдромів. Поряд з новоутвореннями сальних залоз, у цього пацієнта розвинулися новоутворення головного мозку, характерні для синдрому Туркота.[7]
Діагностика
Для діагностики синдрому Муїр-Торре використовують імуногістохімічний метод. Новоутворення сальних залоз зустрічаються нечасто, а імуногістохімія є надійною і легкодоступною, тому дослідники рекомендують її використання. Рутинне імуногістохімічне виявлення білків репарації помилково спарених нуклеотидів допомагає виявити спадковий дефіцит цієї системи.[8]
Лікування
Лікування синдрому Муїр-Торре зазвичай проводитьсяізотретиноїном перорально. Виявлено, що цей препарат запобігає розвитку пухлини.[9][10]
Профілактика
Пацієнти з синдромом Муїр-Торре повинні проходити такий же ретельний скринінг на колоректальну карциному та інші злоякісні захворювання, що й пацієнти з синдромом Лінча. Скринінг складається з частих та ранніх колоноскопій, мамографій, дерматологічної оцінки та візуалізації органів живота й таза.[11]
Епідеміологія
Синдром Муїр-Торре зустрічався у 14 з 50 сімей (28 %) та у 14 із 152 осіб (9,2 %) із синдромом Лінча, також відомим як HNPCC.[12]
Два основні білка репарації помилково спарених нуклеотидів — hMLH1 та hMSH2. Приблизно 70 % пухлин, асоційованих з синдромом Муїр-Торре, мають мікросателітну нестабільність. Хоча спадкове порушення синтезу hMLH1 та hMSH2 зустрічаються рівномірно у HNPCC, порушення синтезу hMSH2 спостерігається у більш ніж 90 % пацієнтів з синдромом Муїр-Торре.[13]
Рак шлунково-кишкового тракту та сечостатевих шляхів — найпоширеніші внутрішні злоякісні новоутворення. Колоректальний рак є найпоширенішим вісцеральним новоутворенням у хворих на синдром Муїр-Торре.[14]
Епонім
Синдром названий іменами Е. Г. Муїр та Д.Торре. Британський лікар Муїр відмітив пацієнта з багатьма кератоакантомами, у якого розвинулось декілька внутрішніх злоякісних новоутворень у молодому віці. Торре представив його висновки на засіданні Нью-Йоркського дерматологічного товариства.[15][16]
З 1980-х років професор Медичної школи Крейтонського університету Генрі Лінч виокремив хворих на синдром Муїр-Торре в сім'ях з синдромом Лінча.[17]
Список літератури
- James, William D.; Berger, Timothy G. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- Association of sebaceous gland tumors and internal malignancy: The Muir–Torre syndrome. The American Journal of Medicine 90 (5): 606–13. May 1991. PMID 2029018. doi:10.1016/S0002-9343(05)80013-0. Проігноровано невідомий параметр
|vauthors=(довідка) - Roberts ME, Riegert-Johnson DL, Thomas BC (2014). A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir–Torre variant of Lynch syndrome. Genetics in Medicine 16 (9): 711–16. PMID 24603434. doi:10.1038/gim.2014.19. Проігноровано невідомий параметр
|doi-access=(довідка) - Roberts ME, Riegert-Johnson DL, Thomas BC (2013). Screening for Muir–Torre syndrome using mismatch repair protein immunohisochemistry of sebaceous neoplasms. J Genet Counsel 22 (3): 393–405. PMID 23212176. doi:10.1007/s10897-012-9552-4.
- Akhtar S, Oza KK, Khan SA (1999). Muir–Torre syndrome: a case report of a patient with concurrent jejunal and ureteral cancer and a review of the literature. J Am Acad Dermatol 41 (5): 681–6. PMID 10534628. doi:10.1016/s0190-9622(99)70001-0.
- Muir–Torre Syndrome / Turcot Syndrome overlap? A patient with sebaceous carcinoma, colon cancer, and a malignant astrocytoma. Dermatology Online Journal 18 (5): 3. May 2012. PMID 22630573. Проігноровано невідомий параметр
|vauthors=(довідка) - Simultaneous Muir–Torre and Turcot's syndrome: A case report and review of the literature. Surg Neurol Int 4 (52): 52. April 2013. PMC 3640225. PMID 23646262. doi:10.4103/2152-7806.110512. Проігноровано невідомий параметр
|vauthors=(довідка) - Towards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient's age or other clinical characteristics. Am J Surg Pathol 33 (6): 934–44. 2009. PMID 19342947. doi:10.1097/PAS.0b013e318199edca. Проігноровано невідомий параметр
|vauthors=(довідка) - Muir–Torre Syndrome – Treatment with Isotretinoin and Interferon Alpha-2a Can Prevent Tumour Development. Dermatology 200 (4): 331–3. March 2000. PMID 10894967. doi:10.1159/000018399. Проігноровано невідомий параметр
|vauthors=(довідка) - Oral isotretinoin therapy for familial Muir–Torre syndrome. J Am Acad Dermatol 12 (3): 475–80. March 1985. PMID 3857234. doi:10.1016/S0190-9622(85)70066-7. Проігноровано невідомий параметр
|vauthors=(довідка) - Ponti G, Pnz de Leon M (2005). Muir–Torre syndrome. Lancet Oncol 6 (12): 980–87. PMID 16321766. doi:10.1016/S1470-2045(05)70465-4.
- The Frequency of Muir–Torre Syndrome Among Lynch Syndrome Families. Oxford Journal 100 (4): 277–81. February 2008. PMID 18270343. doi:10.1093/jnci/djm291. Проігноровано невідомий параметр
|vauthors=(довідка); Проігноровано невідомий параметр|doi-access=(довідка) - Value of MLH1 and MSH2 mutations in the appearance of Muir–Torre syndrome phenotype in HNPCC patients presenting sebaceous gland tumors or keratoacanthomas. |. J Invest Dermatol 126 (10): 2302–7. October 2006. PMID 16826164. doi:10.1038/sj.jid.5700475. Проігноровано невідомий параметр
|vauthors=(довідка); Проігноровано невідомий параметр|doi-access=(довідка) - Muir–Torre syndrome: a case report and review of the literature. Turk J Gastroenterol 23 (4): 394–8. August 2012. PMID 22965514. doi:10.4318/tjg.2012.0411. Проігноровано невідомий параметр
|vauthors=(довідка) - Multiple primary carcinomata of the colon, duodenum, and larynx associated with kerato-acanthomata of the face. Br J Surg 54 (3): 191–5. March 1967. PMID 6020987. doi:10.1002/bjs.1800540309. Проігноровано невідомий параметр
|vauthors=(довідка) - Torre D (November 1968). Multiple sebaceous tumors. Arch Dermatol 98 (5): 549–51. PMID 5684233. doi:10.1001/archderm.98.5.549.
- Lynch HT, Fussaro RM, Roberts L (1985). Muir–Torre syndrome in several members of a family with a variant of the Cancer Family Syndrome. Br J Dermatol 113 (3): 295–301. PMID 4063166. doi:10.1111/j.1365-2133.1985.tb02081.x.