Пріони
Пріони (від англ. proteinaceous infectious particles — білкові заразні частинки) — особливий клас інфекційних патогенів, суто білкових (тобто таких, що не містять нуклеїнових кислот), що спричиняють тяжкі захворювання центральної нервової системи у людей і ряду вищих тварин — пріонові хвороби, що, в свою чергу, входять до групи повільних інфекцій.[1]
? Пріони | ||||
|---|---|---|---|---|
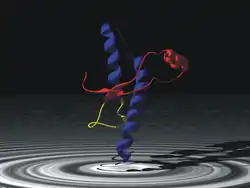 Тривимірна модель пріону | ||||
| Біологічна класифікація | ||||
| ||||
Посилання | ||||
| ||||
- Не плутати з гіпотетичними елементарними частинками — преонами
Пріони здатні збільшувати свою чисельність, використовуючи функції живих клітин (в цьому пріони схожі із вірусами).
Пріонний білок має аномальну тривимірну структуру і здатний прямо каталізувати структурне перетворення гомологічного йому нормального клітинного білка в собі подібний (пріоновий), приєднуючись до білка-мішені і змінюючи його конформацію. Як правило, пріонний стан білка характеризується переходом α-спіралей білка в β-складчатість.
Історія
У другій половині XX століття лікарі зіткнулися із незвичайним захворюванням людини — поступовим прогресуючим руйнуванням головного мозку, що відбувається у результаті загибелі нервових клітин. Це захворювання отримало назву губчастої енцефалопатії. Схожі симптоми були відомі давно, але вони спостерігалися не в людини, а у тварин (скрепі овець), і довгий час між ними не знаходили достатнього обґрунтованого зв'язку.
Новий інтерес до їх вивчення виник в 1996 р., коли у Великій Британії з'явилася нова форма захворювання, що позначається як «новий варіант хвороби Кройцфельда-Якоба (nvCJD)».
Важливою подією було поширення «коров'ячого сказу» у Великій Британії, епідемія якого була початково в 1992—1993 рр., а потім і в 2001 р., та охопила кілька європейських держав. Тим не менше м'ясо корів було експортовано в багато країн. Захворювання пов'язують із використанням «пріонізованого» кісткового борошна в кормах і преміксах, що було виготовлено із туш полеглих або хворих тварин, які можливо, і не мали явних ознак захворювання.
Шляхи перенесення фактора, що спричиняє хворобу, механізми проникнення пріонів в організм і патогенез захворювання вивчені поки недостатньо.
У 1997 р. американському науковцю Стенлі Прузінеру була присуджена Нобелівська премія з фізіології або медицини за вивчення пріонів.
Властивості молекул
Пріонові білки ссавців не подібні до пріонового білка дріжджів по амінокислотній послідовності. Попри це, основні структурні особливості (формування амілоїдних волокон і їх висока специфічність, що перешкоджає передачі пріонів від одного виду організмів до іншого) у них спільні. Водночас, пріонний білок, що відповідає за коров'ячий сказ, має здатність передаватися від виду до виду.
Правий малюнок — модель двох конформацій пріона; ліворуч відома, нормальна, конформація структури термінального ділянки C-terminal PrP C .[2]
Молекулярні основи патогенезу

У ході досліджень мозкових тканин померлих від пріонних інфекцій тварин було показано, що пріони не містять нуклеїнових кислот, а являють собою білки. Одним із перших детально охарактеризованих пріоннних білків став PrP (від англ. prion-relatedprotein або protease-resistantprotein) масою близько 35 к Да. Відомо, що PrP може існувати в двох конформаціях — «здоровій» — PrP C , яку він має в нормальних клітинах (C- від англ. cellular — «клітинний»), в якій переважають альфа-спіралі, і «патологічній» — PrP Sc , власне пріонній (Sc - від англ. scrapie), для якої характерна наявність великої кількості бета-тяжів. При потраплянні в здорову клітину, PrP Sc каталізує перехід клітинного PrP C у пріонну конформацію. Накопичення пріонного білка супроводжується його агрегацією, утворенням високовпорядкованих фібрил (амілоїду), що зрештою призводить до загибелі клітини. Пріони, що вивільнилися, мабуть, виявляються здатними проникати в сусідні клітини, також викликаючи їх зараження і загибель.
Функції білка PrP C у здоровій клітині поки що точно не визначені. У нормі білок PrP C асоційований із клітинною мембраною, глікозильований залишком сіалової кислоти. Він робить циклічні переходи всередину клітини і назад на поверхню в ході ендо — та екзоцитозу . Один такий цикл триває близько години. У ендоцитозному пухирці або на поверхні клітини молекула PrP C може розрізатися протеазами на дві приблизно рівні частини.
До кінця механізм спонтанного виникнення пріонних інфекцій не з'ясований. Вважається (але ще не повністю доведено), що пріони утворюються в результаті помилок у біосинтезі білків. Мутації генів, що кодують пріонний білок (PrP), помилки трансляції, процеси протеолізу — вважаються головними кандидатами на механізм виникнення пріонів.
Є дані, що дають підставу вважати, що пріони є не тільки інфекційними агентами, але й мають функції в нормальних біопроцесах. Так, наприклад, існує гіпотеза, що через пріони здійснюється механізм генетично обумовленого стохастичного старіння.
Класифікація
| Пріони ссавців — збудники губчастої енцефалопатії | ||||
|---|---|---|---|---|
| ICTVdb Code | Захворювання | Носій | Назва пріона | PrP ізоформа |
| 90.001.0.01.001. | Скрепі | Вівці та кози | Пріони скрепі | OvPrP Sc |
| 90.001.0.01.002. | Трансмісивна енцефаломіопатія норок (ТЕН) | Норки | Пріони ТЕН | MkPrP Sc |
| 90.001.0.01.003. | Chronic wasting disease (CWD) | Олені та лосі | CWD пріони | MDePrP Sc |
| 90.001.0.01.004. | Губчаста енцефалопатія великої рогатої худоби (ГЕВРХ) | Корови | Пріони ГЕВРХ | BovPrP Sc |
| 90.001.0.01.005. | Губчаста енцефалопатія котячих (ГЕК) | Кішки | Пріони ГЕК | FePrP Sc |
| 90.001.0.01.006. | Exotic ungulate encephalopathy (EUE) | Nyala and greater kudu | Пріони | NyaPrP Sc |
| 90.001.0.01.007. | Куру | Люди | Пріони куру | HuPrP Sc |
| 90.001.0.01.008. | Хвороба Кройцфельда — Якоба (ХКЯ) | Люди | Пріони ХКЯ | HuPrP Sc |
| (New) Variant Creutzfeldt-Jakob disease (vCJD, nvCJD) | Люди | VCJD пріони | HuPrP Sc | |
| 90.001.0.01.009. | Синдром Герстмана—Штройслера—Шейнкера (GSS) | Люди | GSS пріони | HuPrP Sc |
| 90.001.0.01.010. | Фатальне сімейне безсоння (ФСБ) | Люди | Пріони ХСБ | HuPrP Sc |
Етіологія
Людина може заразитися пріонами, що містяться в їжі, оскільки вони не руйнуються ферментами травного тракту. [джерело?] Безперешкодно проникаючи через стінку тонкого кишечника, вони в кінцевому результаті потрапляють в центральну нервову систему. Так переноситься новий варіант хвороби Кройцфельда — Якоба (nvCJD), якою люди заражаються після вживання в їжу яловичини, що містить нервову тканину із голів худоби, яка хвора бичачою губчастою енцефалопатією (BSE, коров'ячий сказ).
Пріони можуть проникати в тіло і парентеральним шляхом. Були описані випадки [джерело?] зараження при внутрішньом'язовому введенні препаратів, виготовлених із людських гіпофізів (головним чином гормон росту для лікування карликовості), а також зараження мозку інструментами при нейрохірургічних операціях, оскільки пріони стійкі до вживаних в наш час[коли?] термічних та хімічних методів стерилізації. Ця форма хвороби Крейтцфельдта-Якоба позначається як ятрогенна (1CJD).
Згідно із загальноприйнятих теорій, при досі нез'ясованих умовах, в організмі людини може відбутися спонтанна трансформація «потенційно» пріонного протеїну в «активну пріонну частинку». Так виникає так звана спорадична хвороба Кройтцфельда-Якоба (sCJD), вперше описана в 1920 р. незалежно один від одного Гансом Герхардом Кройцфельдом і Альфонсом Марією Якобом. Передбачається, що спонтанне виникнення цієї хвороби пов'язане із фактом, що в нормі в людському тілі постійно виникає невелика кількість пріонів, які ефективно ліквідуються клітинним апаратом Гольджі. Порушення цієї здатності «самоочищення» клітин може призвести до підвищення рівня пріонів вище допустимої межі норми і до їх подальшого неконтрольованого розповсюдження. Причиною виникнення спорадичної хвороби Кройцфельда-Якоба згідно з цією теорією є порушення функції апарату Гольджі в клітинах.
Особливу групу пріонних захворювань являють собою спадкові (вроджені) хвороби, викликані мутацією гена пріонного протеїну, який робить пріонний протеїн, що утворився, вразливішим до спонтанної зміни просторової конфігурації та перетворення їх у пріони. До цієї групи спадкових захворювань відноситься і спадкова форма хвороби Кройцфельда-Якоба (fCJD), яка спостерігається в ряді країн світу.
При пріонових патологіях найвища концентрація пріонів виявлена в нервовій тканині заражених людей. Значна кількість пріонів зустрічається в лімфатичній тканині. Наявність пріонів в біологічних рідинах, включаючи слину, поки що не була однозначно підтверджена. Якщо уявлення про постійне виникнення невеликої кількості пріонів правдиві, то можна припустити, що нові, чутливіші методи діагностики відкриють цю кількість пріонів, що розкидані по різних тканинах. У цьому випадку, однак, мова піде про «фізіологічні» рівні пріонів, які не являють собою ніякої загрози для людини.
Шляхи зараження
[джерело?]
Дуже мало відомо про молекулярний характер пріонів, що спричинюють захворювання. Зараження може бути при потраплянні приблизно 100 000 молекул, які в більшості випадків створюють великі скупчення. Значення агрегації окремих молекул в асоціації для вірулентності пріонів поки що не відома. Не можна виключити, що вірулентними є і окремі молекули пріонів. З деяких експериментів випливає, що для виникнення пріонів в тканині достатньо лише тимчасового контакту тканини з матеріалом, що містить пріони, і немає необхідності, щоб пріони були назавжди внесені в організм. Цей ризик є актуальним, наприклад, у зв'язку із використанням хірургічних інструментів, заражених пріонами. Процес трансформації «здорових» пріонних протеїнів у пріони може бути ініційований простим контактом здорових тканин із пріонами, зафіксованими на хірургічному інструменті.
Перебіг хвороби і поширення пріонів по організму залежить від типу пріона. Пріони відрізняються складом амінокислот, характерних для даного виду. Це визначається видовим геном пріонового протеїну, а також так званими посттрансляційними модифікаціями або ступенем глікозилювання базового білкового ланцюжка. Посттрансляційна модифікація значно впливає на характеристики пріонів і саме їй приписують різницю між так званими пріоновими родами. У випадку нового варіанту (nvCJD) був поки що описаний лише один вид пріона, схожий із пріонами худоби, зараженої бичачою губчастою енцефалопатією. Тому перебіг захворювання у людини і тварин, заражених новими варіантами, практично однаковий. У інших видів живих істот, однак, відомо багато пріонових родів. У овець було описано приблизно два десятки таких родів, які не вірулентні для людини. Перебіг овечого пріонового захворювання, залежно від роду пріонів, — драматично відрізняється — від дуже швидкого, з практично раптовою загибеллю, до повільного, затяжного.
Нетипові випадки клінічного перебігу нового варіанту у худоби, зараженої бичачою губчастою енцефалопатією, які траплялися в Японії та Італії [джерело?], наводять на думку про існування більшої кількості родів бичачих пріонів. Якщо б цей рід бичачих пріонів потрапив в організм людини, слід було б очікувати виникнення нового варіанту із симптомами і клінічним перебігом, відмінними від відомих випадків.
У пацієнтів, хворих на хворобу Кройцфельда-Якоба, пріони поширюються у нервовій системі, тканинах ока і лімфатичних тканинах, включаючи мигдалини, селезінку, а також у сліпій кишці. Найбільша кількість пріонів знаходиться в нервовій системі, а найменше — в лімфатичній тканині.
Поки що не було зареєстровано жодного випадку перенесення нового варіанту хвороби Кройцфельда-Якоба (nvCJD) при медичному втручанні, що є, зрозуміло, гарною новиною. З іншого боку, фахівці попереджають про перебільшений оптимізм, перш за все в умовах Великої Британії, оскільки інкубаційний період може бути досить тривалим (від 5-8 місяців до 10-15 років). [джерело?]
Пріони та медичні інструменти
[джерело?] Пріони дуже стійкі до звичайних методів дезінфекції. Іонізуюче, ультрафіолетове або мікрохвильове випромінювання на них практично не діє. Дезінфекційні засоби, що зазвичай використовуються в медичній практиці, діють на них лише в дуже обмеженій формі. Надійно їх ліквідовують дезінфікуючі реактиви — сильні окисники, що мають руйнівну дію на протеїни .[джерело?]
Інше утруднення являє собою стійкість пріонів до високих температур. Навіть при автоклавуванні при 134 ° C протягом 18 хвилин неможливо досягти повного руйнування пріонів, і пріони «виживають» у формі, здатній викликати зараження. Стійкість до високих температур ще більше зростає, якщо пріони засохнуть на поверхні металу або скла, або якщо зразки перед автоклавуванням були піддані дії формальдегіду. [джерело?]
У Великій Британії, де новий варіант є дуже серйозною проблемою, із цих причин вже використовуються одноразові хірургічні інструменти для тонзилектомії. У майбутньому напрошується альтернативне рішення: створення нових інструментів, із урахуванням підвищених вимог до очищення та знезараження. Одноразове використання інструментів згідно з принципами ВООЗ потрібно в разі стоматологічного обслуговування пацієнтів з діагностованим пріонним захворюванням або у випадку підозри на нього.
Набагато складнішим рішенням цієї проблеми є лікування пацієнтів групи ризику. До них належать пацієнти, які зазнали операцій, при яких була використана потенційно заражена тверда мозкова оболона, або пацієнти з сімей із спадковою формою хвороби Кройцфельда-Якоба. ВООЗ у цьому випадку не вимагає ніяких спеціальних заходів. Британський Консультаційний науковий комітет із губчастої енцефалопатії у своєму рішенні в 1998 р. визнав за можливе обмежитися ретельнішим очищенням та знезараженням інструментів, у поєднанні з тривалішим автоклавуванням.
Пріонові хвороби людини
- Хвороба Кройцфельда — Якоба (Creutzfeldt-Jakob disease);
- Фатальне сімейне безсоння (Fatal Familial Insomnia);
- Куру (Kuru);
- Синдром Герстмана—Штройслера—Шейнкера (Gerstmann-Sträussler-Scheinker disease).
Потенційна небезпека для людини
Незважаючи на невелику кількість явних випадків пріонних захворювань у людей, багато фахівців вважають, що існує високий ступінь небезпеки їх для людини.
За неперевіреними даними, джерелом поширення можуть бути стоматологічні процедури, пов'язані з попаданням пріонів в кров'яне русло.
Під підозру потрапив також лецитин тваринного походження, що викликало скорочення застосування його в фармакологічній промисловості, і витіснення рослинним (в основному, соєвим) лецитином.
Дослідження пріонів дріжджів та інших мікроміцетів
Пріон-подібні білки, поведінка яких подібна до поведінки PrP, знайдені в природних популяціях дріжджів та інших мікроміцетів. Дослідження пріонів дріжджів підтвердили гіпотезу про те, що перетворення білків у пріоновий стан залежить тільки від білків. Було показано, що пріони, екстраговані з клітин, можуть служити «зерном» утворення пріонів у пробірці. Один із найґрунтовніше досліджених білків, схильних до утворення пріонів у дріжджів — фактор термінації трансляції (eRF3), який утворює так названі PSI + клітини. Такі клітини мають змінений фізіологічний стан і змінений рівень експресії деяких генів, що дозволило висунути гіпотезу про те, що у дріжджів утворення пріонів може грати адаптивну роль[3].
Критика загальноприйнятих теорій щодо етіології пріонових хвороб
Марк Парді та Девід Браун припустили, що металлоіони, взаємодіючи з протеїнами пріона, можуть бути причиною розвитку пріон-індукованих (prion-mediated) захворювань.[4]
Парді провів кластерне епідеміологічне дослідження при пріонних захворюваннях в районах з низькою концентрацією міді в ґрунтах. [джерело?]
Примітки
- Somerville RA. (2002). TSE agent strains and PrP: reconciling structure and function. Trends in Biochemical Sciences 27 (12): 606–612 year=2002. PMID 12468229.
- для відображення завантаження див. RCSB Protein Databank
- Galkin AP, Mironova LN, Zhuravleva GA, Inge-Vechtomov SG. (2006). Yeast prions, mammalian amyloidoses, and the problem of proteomic networks. Genetica 42 (11): 1558. PMID 17163073.
- 2000 — 09-22, Normal Function of Prions, Statement to the BSE Inquiry. Архів оригіналу за 16 грудня 2008. Процитовано 18 вересня 2009.
Див. також
Джерела
- І. С. Шкундіна, М. Д. Тер-Аванесян. Пріони. Успіхи біологічної хімії, т. 46, 2006 (огляд)
- Григор'єв В. Б. — Пріонні хвороби людини і тварин. — Питання вірусології, Т.49 (№ 5), с.4-12, 2004 (огляд)
- Ing. Jaroslav Petr, DrSc. Phony a ustni dutina. Progresdent, 2004, № 2, s. 12-16
Посилання
- ПРІОНИ
- Mad Cow Disease Інформація про коров'ячий сказ, Center for Global Food Issues. (англ.)
- Madcowering A BSE-TSE blog. (англ.)
- The Pathological Protein — Mad Cow, Chronic Wasting, and Other Deadly Prion Diseases (2003, updated online 2005). Philip Yam, Scientific American magazine writer and News Editor. (англ.)
- пріонні захворювання (2003). Dr. Sean Heaphy, Leicester University. (англ.)
- Prion Diseases and the BSE Crisis (1997). Стаття Stanley Prusiner — першовідкривача пріонів, з Science magazine. (англ.)
- Britannica Nobel: пріони, 1997 (англ.)
- ICTVdb 90.001.0.01. Mammalian Prions (англ.)
- Офіційна сторінка сайту про хвороби коров'ячого сказу Mad Cow Disease (англ.)
- News & Views on Mad Cow Disease, Mad Deer Disease, Chronic Wasting Disease, and Bovine Spongiform Encephalopathy (англ.)
- Biography of Dr Prusiner (англ.)
- Science Daily Стаття про вакцину проти пріонних захворювань (англ.)
- Science Daily article on transmission of prions through soil (англ.)
- Хороший огляд по пріонами з Science Creative Quarterly (англ.)
- Пріони — новий клас збудників інфекційних захворювань (рос.)