Пріонові хвороби
Пріо́нні або пріо́нові хворо́би (англ. Prion-related diseases, нерідко використовують ще й термін англ. Transmissible spongiform encephalopathy — трансмісивна губчаста/спонгіформна енцефалопатія) — група нейродегенеративних захворювань людини і тварин з утворенням губчастої енцефалопатії, які належать також до групи повільних інфекцій і характеризуються ураженням центральної нервової системи (ЦНС), м'язової, лімфоїдної та інших систем, завжди закінчуються смертю.
| Пріонові хвороби | |
|---|---|
| Спеціальність | інфекційні хвороби |
| Причини | пріон |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | 8E02 |
| МКХ-10 | A81[1] |
| DiseasesDB | 25165 |
| eMedicine | neuro/662 |
| MeSH | D017096 |
| | |
Збудником цих хвороб є пріони — винятковий клас інфекційних агентів білкової природи, які не містять у своїй структурі нуклеїнових кислот і тим самим відрізняються від інших збудників інфекційних хвороб.
На сьогодні встановлено, що крім короткоживучих білків, які організм використовує як енергетичний ресурс і як будівельний матеріал для нових клітин, є й такі білки, яким притаманні зовсім інші, й досі, значною мірою, непрояснені функції. Саме з ними і пов'язують виникнення повільних пріонових інфекцій. Виходячи з того, що ці хвороби інфекційної природи, а інфекційний агент передається від джерела до сприйнятливого організму, експерти ВООЗ вживають поняття збудник по відношенню до пріонів.
Нині до пріонових хвороб відносять:
- у тварин скрепі, губчасту енцефалопатію великої рогатої худоби (коров'ячий сказ), хронічну виснажливу хворобу диких копитних в неволі тощо.
- у людей до пріонових хвороб долучають куру, хворобу Кройцфельда-Якоба, синдром Герстмана—Штройслера—Шейнкера, фатальне сімейне безсоння.
Є припущення, що патогенні пріони мають відношення до спричинення ще деяких хвороб людей: хронічної прогресуючої енцефалопатії дитячого віку / хвороби Альперса, спонгіформного міозиту з пріон-асоційованими включеннями. При них у тканинах знаходять амілоїдні зміни і скупчення нормальних пріонів, хоча досі патогенні пріони не були знайдені.

Історичні відомості
У 1920 році німецький невролог Г-Ґ. Кройцфельд та в 1921 році ще один німецький невролог А. Якоб незалежно один від одного описали симптоми хвороби, яка в подальшому була визнаною першим пріоновим захворюванням людини, отримала назву за їхніми прізвищами і була долучена в подальшому до переліку пріонових хвороб. У 1954 році ісландський вчений Б'єрн Сігурдссон виклав результати свої багаторічних досліджень скрепі — хвороб овець у Ісландії, які проявлялися порушенням координації рухів, паралічами, нестерпною сверблячкою і завжди завершувалися смертю, вперше відніс їх до групи повільних інфекцій та привернув увагу до таких недуг з характерним патоморфологічним субстратом — губчастою енцефалопатією. Протягом багатьох років американський вірусолог Деніел Карлтон Гайдушек вивчав загадкову хворобу куру і в 1965 році сповістив медичну спільноту, що вона є інфекційною і йому вдалося це довести. У 1982 році американський біохімік Стенлі Прузінер успішно довів, що біологічною структурною основою так званого «невстановленого трансмісивного агента» скрепі є патологічна (інфекційна) ізоформа клітинного протеїну Р (або РrРsc[2]). Науковець створив новий термін «prion» (використавши сполучення букв від англ. proteinaceous infectious particles — «протеїнові інфекційні частки») для того, щоб підкреслити специфіку інфекційного агента, що принципово відрізняється від вірусів і віроїдів своїми біологічними характеристиками[3].
Актуальність
Актуальність проблеми пріонових хвороб зумовлена тим, що вони надзвичайно небезпечні для життя людей і тварин. Ці недуги виявляються у всіх країнах світу, а збудники скрепі та коров'ячого сказу з Великої Британії завезені в ряд країн Європи, Близького Сходу, Азії. Сучасні карантинні заходи для ліквідації епізоотії трудомісткі, завдають величезних економічних збитків сільському господарству, а також мають негативний вплив на соціальну ситуацію. Двом дослідникам проблеми пріонових хвороб — К. Гайдушеку (1976 р.) та С. Прузінеру (1997 р.) була присуджена Нобелівська премія з медицини.
Етіологія
Збудниками пріонових хвороб є патологічні ізоформи пріонів і змінені білкові молекули хазяїна, які не мають нуклеїнової кислоти, але характеризуються патогенними властивостями. Пріони складаються тільки зі змінених (конформаційних) білкових молекул. Відсутність у складі пріонів нуклеїнових кислот визначає незвичність деяких з властивостей. Вони дуже стійкі у довкіллі — з усього живого пріони гинуть останніми: витримують кип'ятіння протягом 30-60 хвилин, висушування, заморожування, ультрафіолетове і гамма-опромінення, хімічну обробку спиртами, формальдегідами, кислотами, не піддаються гідролізу ферментами. Ген, який кодує пріоновий білок, міститься не у складі пріону, а в клітині. Пріоновий білок, потрапляючи в організм, активує цей ген PrNP і зумовлює індукцію синтезу аналогічного білка. Разом з цим пріони при всій своїй структурній і біологічній своєрідності мають ряд властивостей звичайних вірусів (віріонів). Вони проходять крізь бактерійні фільтри, не розмножуються на штучних живильних середовищах, репродукуються до концентрацій 105−1011 на 1 г мозкової тканини, адаптуються до нового хазяїна, змінюють патогенність і вірулентність, відтворюють феномен інтерференції, характеризуються штамовими відмінностями, здатністю до персистенції в культурі клітин, отриманих з органів зараженого організму, можуть бути клоновані.
Детальніші відомості з цієї теми ви можете знайти в статті Пріони.
Епідеміологічні особливості
Джерело і резервуар інфекції
У більшості захворілих патологічний пріон PrPsc з'являється спонтанно — шляхом мутації власного PrPc. В інших випадках можливе екзогенне зараження від тварин, які є основним джерелом збудника (вівці, кози, корови, олені та інші копитні тварини, норки, котячі, а, можливо, й інші види). Лише при куру джерелом збудника в природі вважають людину — хвору або ту, яка перебуває в інкубаційному періоді недуги[4].
Механізм і фактори передачі
Пріонові хвороби успадковуються за автосомно-домінантним типом (щоправда, це непрямий процес — через попередню генну автореплікацію інфекційного агента). Хвора людина в побутових умовах не становить небезпеки для оточуючих. Однак концентрація пріонів у її органах в інкубаційному періоді значна, і це створює небезпеку зараження реципієнтів від інфікованих, але ще не хворих донорів крові або внутрішніх органів, ятрогенним[5] шляхом (інтрацеребрально через пересадку рогівки, фрагментів твердої мозкової оболони, імплантацію внутрішньочерепних електродів, внутрішньовенно, інтраперитонеально, через шкіру[6], при проведенні ендоскопії[7] тощо). Найбільша концентрація збудника в мозковій тканині, але він у значних титрах виявляється в тканинах інших органів, у крові[8]. Хвора людина в домашніх побутових умовах або в лікарні не створює небезпеки для оточуючих, якщо вони не контактують з тканинами, особливо мозковою, ураженої людини. Враховуючи те, що концентрація пріонів в органах у період інкубації значна, а сам період тривалий, людина може стати джерелом збудника при переливанні крові, при трансплантації органів, взятих від заражених, але поки що не захворілих[9]. Тварини заражаються або за рахунок спонтанних мутацій, або внаслідок поїдання заражених тварин.

Механізми передачі пріонів різноманітні й включають у природі аліментарну передачу, гіпотетично можлива й гемоконтактна і аерозольна передача[10], можливий також вертикальний шлях передачі через плаценту. Особливу епідемічну небезпеку становить така форма перебігу повільних інфекцій (наприклад, при скрепі, коров'ячому сказі тощо), при якій приховане вірусоносійство і типові морфологічні зміни в організмі перебігають безсимптомно. Передача пріонів можлива у разі споживання м'яса хворих тварин або, частіше, тих, що були у періоді інкубації, коли у них будь-яких симптомів хвороби не спостерігається; а також під час розтину загиблих тварин. Враховуючи високу терморезистентність пріонів, звичайна термічна кулінарна обробка м'яса не знешкоджує збудника хвороби. Зараження може статись і при парентеральному введенні гормонів, виготовлених з органів інфікованих тварин (пітуітрин) чи людей (гонадотропін). На небезпеку інфікування наражаються також працівники боєнь, м'ясокомбінатів під час забою тварин в інкубаційному періоді й контакту з їх тканинами та органами. Особливості епідемічного процесу серед споживачів м'яса інфікованих тварин вивчені недостатньо, а сам факт зараження людей від корів доведений лише на початку 90-х років XX століття.
Сприйнятливий контингент та імунітет
Особливо уразливими вважають реципієнтів трансплантації внутрішніх органів, тих осіб, що піддаються різним медичним маніпуляціям, при яких прилади чи механізми можуть бути контаміновані тканинами заражених донорів, отримувачі деяких медичних препаратів, що містять гормони, ветеринарні й медичні хірурги, патоморфологи, ветеринари, працівники м'ясопереробної промисловості тощо. Імунітет внаслідок захворювання не виробляється, пріонні хвороби завжди закінчуються смертю.
Патогенез
Загальні риси
Загальною патогенетичною основою повільних інфекцій є накопичення збудника в різних органах і тканинах зараженого організму задовго до перших клінічних проявів і тривала, іноді багаторічна, персистенція в тих органах, в яких тривалий час не виявляють патогістологічних змін. Згодом відбувається проліферація різних клітин. Наприклад, губкоподібні енцефалопатії характеризуються вираженим гліозом, патологічною проліферацією і гіпертрофією астроцитів, що і спричиняє вакуолізацію і загибель нейронів, тобто розвиток губкоподібного стану тканини мозку[11]. Інкубаційний період при усіх пріонових хворобах дуже тривалий — від декількох місяців до десятиліть.
Патоморфологічні зміни
Ці зміни при пріонових інфекціях можна підрозділити на ряд характерних процесів, серед яких перш за все слід назвати дегенеративні зміни в ЦНС. Таким чином, будучи не антигенними, пріони спричинюють не запальні, а дегенеративні процеси[12]. Зміна конститутивних білків — нормальних клітинних пріон-протеїнів — PrPc за розмірами і/або формою призводить до їх перетворення з життєво необхідних до смертельно небезпечних — PrPsc. Цей феномен пропонується називати терміном «конформаційні хвороби». Є підстави вважати, що такі конформаційні білки можуть виконувати роль головних регуляторів в організмі, у тому числі й такого найважливішого процесу як обмеження тривалості самого життя. Після проникнення у клітину PrPsc стає матрицею для перетворення (конвертації) інших молекул PrPc у PrPsc, активації гену PrNP[13]. Доведено, що у мозку хворих зв'язані з ліпопротеїнами низької і дуже низької щільності[14]Акумуляція PrPsc у клітинах ЦНС призводить до їх незворотної дегенерації і загибелі, через що пріонові хвороби абсолютно смертельні.
Клінічні прояви
В діючий на сьогодні МКХ 10-го перегляду у класі «Деякі інфекційні та паразитарні хвороби», підкласі «Вірусні інфекції ЦНС» під кодом А81 включені «Атипові вірусні інфекції ЦНС», до яких залучено «Хворобу Кройцфельда-Якоба» (А81.0), «Інші атипові вірусні інфекції ЦНС» (А81.8), куди рекомендують шифрувати куру, та «Атипові вірусні інфекції ЦНС не уточнені» (А81.9), куди рекомендують шифрувати всі інші пріонові хвороби. Ця класифікація була впроваджена у 1993 році, тому й відображає застарілі поняття про пріонові хвороби, які не є вірусними. На 2015 рік планується впровадження МКХ 11-го перегляду, де ці класифікаційні моменти будуть виправлені.
Розрізняють три класичні форми пріонових спонгіформних енцефалопатій:
- спорадичну (більшість від усіх випадків),
- сімейну або спадкову (близько 10-15%),
- ятрогенну (відсоток таких випадків ще остаточно не встановлений).
Відповідно у людей за механізмом розвитку розрізняють такі пріонові хвороби:
- спорадичні (спорадична й аміотрофічна форми хвороби Кройцфельда-Якоба),
- спадкові (спадкова форма хвороби Кройцфельда-Якоба, синдром Герстмана-Штройслера-Шейнкера, фатальне сімейне безсоння)
- набуті (куру, ятрогенна форма та новий варіант хвороби Кройцфельда-Якоба).
У більшості випадків повільні інфекції виникають і розвиваються без температурної реакції організму. Всі підгострі трансмісивні губкоподібні енцефалопатії проявляються порушеннями ходи і координації рухів. Нерідко ці симптоми є найбільш ранніми, пізніше до них приєднуються геміпарези й паралічі. Усі форми пріонової енцефалопатії проявляються різноманітною неврологічною й психічною симптоматикою. Передусім це:
- розлади чутливої сфери (амнезія різного ступеня, втрата й спотворення чутливості, випадання функцій органів чуттів);
- ураження рухової сфери (атаксія, адинамія, атрофія м'язів, парези й паралічі);
- порушення психіки (втрата фахових навичок, депресія, сонливість, агресивність, зниження інтелекту аж до повного недоумства)[15].
Здебільшого при всіх пріонових хворобах хворі гинуть від виснаження або приєднання пневмонії.
Детальніші відомості з цієї теми ви можете знайти в статті Куру (хвороба).
Детальніші відомості з цієї теми ви можете знайти в статті Хвороба Кройцфельда-Якоба.
Детальніші відомості з цієї теми ви можете знайти в статті синдром Герстмана—Штройслера—Шейнкера.
Детальніші відомості з цієї теми ви можете знайти в статті фатальне сімейне безсоння.
Діагностика
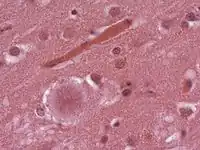
Рутинні параклінічні методи малоінформативні. Досить серйозну діагностичну підтримку надає МРТ ЦНС. Вона показує гіперінтенсивні сигнали в корі головного мозку, базальних гангліях та таламусі. Описані характерні зміни у вигляді «хокейної ключки» — збільшення інтенсивності сигналу у шкарлупі та центрі хвостатого тіла нагадують цей предмет. Так само характерним є симптом «медових сотів» — відповідне за картиною збільшення інтенсивності сигналу у таламусі.
Одним з досить чутливих специфічних експрес-методів діагностики пріонових захворювань є біопсія лімфатичного вузла і забарвлення матеріалу конго-червоним з метою виявлення амілоїдозу. За наявності амілоїдозу можна експериментально заразити мавп або мишей суспензією, приготовленою з мозку чи лімфатичних вузлів, взятих від померлих людей або тварин. З 4-14-го тижня захворювання пріони можна виявити у спинному, головному мозку та інших органах. Пріон можна виявити у біоптаті глоткового мигдалика, а також при біопсії мозку з імуногістохімічним типуванням протеїнів. При гістопатологічному вивченні ЦНС знаходять спонгіоз з вакуолізацією нейронів, проліферацію астроцитів і глії без ознак запалення та демієлінізації, амілоїдні пріон-протеїнові бляшки. У разі пріон-асоційованого міозиту з включеннями гістологічно виявляється некротична міопатія з наявністю вакуоль, які в заморожених зрізах містять спіралеподібні конгофільні нитки.
Для встановлення точного діагнозу пріонового захворювання людини потрібно виявити один з чотирьох додаткових критеріїв:
- наявність специфічних амілоїдних бляшок, спричинених пріонами (Pr-амілоїдних бляшок);
- спроможність тканини до зараження спонгіформною енцефалопатією тварин;
- наявність ізоформ пріон-протеїну Кройцфельда-Якоба;
- наявність патогенного мутованого гена PrNP[16].
У лікворі хворих виявляють білок, який може служити діагностичним тестом спонгіформної енцефалопатії людей і тварин. Перспективним є тест з стрептоміцином, який виявляє пріон у різних тканинах.
Лікування
Ефективну етіотропну і патогенетичну терапію пріонових хвороб не розроблено. У ранніх стадіях використовують симптоматичні засоби, що коригують порушення поведінки, розлади сну і міоклонію (амфетаміни, барбітурати, антидепресанти, бензодіазепіни, інші нейролептики); у пізніх — підтримуючу терапію.
Припускають, що препарати, які зв'язують і стабілізують структуру клітинної ізоформи пріону, можуть зменшувати експресію пріонів і затримувати початок хвороби. Ведеться пошук хімічних агентів, які б дестабілізували структуру патологічної ізоформи пріону. Хімічні впливи на ендоцитоз, екзоцитоз, внутрішньоклітинний транспорт і специфічне руйнування пріонів також можуть виявитися ефективними. Однак досі прогноз при пріонових інфекціях завжди несприятливий.
Профілактика
У людей
Засобів специфічної профілактики не існує. При роботі з хворими у процесі інвазивних процедур, а також при контакті з їх біологічними рідинами необхідно дотримуватися правил, передбачених при роботі з патогеном особливого ступеня небезпеки (як при ВІЛ-інфекції), використовуючи спеціальний захисний одяг (рукавички з металевим прошарком, маску, захисні окуляри, фартух). Це також стосується осіб, котрі контактують з потенційно можливими джерелами інфекційних пріонів і належать до груп ризику (ветеринарні й медичні хірурги, патоморфологи, ветеринари, працівники м'ясопереробної промисловості тощо). Трупи померлих від пріонових хвороб повинні підлягати кремації.
Найбільш реальні три методи знезаражування забрудненого пріонами медичного інструментарію:
- автоклавування при 134–138 °C протягом 18 хвилин (у разі неповного завантаження) або при 132°Cпротягом 60 хв. (у випадку використання горизонтального автоклаву);
- обробка гідроксидом натрію (віддаючи перевагу концентрації 1 моль/л і експозиції 60 хв. при 20 °C);
- обробка гіпохлорітом натрію (бажано розчином з вмістом 2,5% активного хлору протягом 60 хвилин при 20 °C).
Надалі проводиться контроль якості проведеного знезараження[17] Також застосовується обмеження використання лікарських засобів, виготовлених з тканин корів, припинене виробництво гормональних препаратів гіпофізу тваринного й людського походження, посилені вимоги до їх сертифікації, перевага надається генно-інженерним препаратам. Введено обмеження на трансплантацію твердої мозкової оболони, рогівки. Заборонено трансплантацію тканин, переливання крові й призначення препаратів крові від осіб з нерозшифрованою деменцією.
У тварин
При інфекційних амілоїдозах тварин надзвичайно важливе значення відіграє епізоотологічний нагляд, який включає недопущення завозу збудника з імпортованими інфікованими тваринами, з натуральним м'ясом і, навіть, кулінарно обробленими м'ясними продуктами. Щоб обмежити поширення збудника серед тварин, необхідно якомога раніше виявити хворих і знищити (з невідкладною кремацією) не тільки їх, але й усе стадо, в якому була хвора тварина. Категорично заборонено додавати в корм худобі білкові концентрати тваринного походження. Ще одним шляхом запобігання пріоновим хворобам може бути розведення домашніх тварин, генетично резистентних до пріонів.
Примітки
- Згідно з МКХ-11, яка пропонується на зміну МКХ-10, пріонові хвороби класифікуються як 8E00-8E0Z
- «sc» від «scraepie» — пріонове захворювання овець).
- Prusiner S. Autobiography. Nobel Prize in Physiology or Medicine 1997. .
- McAlister V. Sacred disease of our times: failure of the infectious disease model of spongiform encephalopathy. Сlin. Invest. Med. June 2005. — issue 3. — р.101-4
- Тобто спричинена медичними втручаннями.
- Gwennaelle J. Wathne, Adrien Kissenpfennig, Bernard Malissen, Chiara Zurzolo and Neil A. Mabbott Determining the role of mononuclear phagocytes in prion neuroinvasion from the skin. Journal of Leukocyte Biology vol. 91 no. 5 817-828 [недоступне посилання з липня 2019]
- M W Head, J W Ironside vCJD and the gut: implications for endoscopy. Gut 2007;56:9-11 doi:10.1136/gut.2006.101964
- Brown P, Preece M, Brandel JP, Sato T, McShane L, Zerr I, Fletcher A, Will RG, Pocchiari M, Cashman NR, d'Aignaux JH, Cervenakova L, Fradkin J, Schonberger LB, Collins SJ Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000. — volume.55, issue № 8. — pages 1075-81.
- Nora Hunter, James Foster, Angela Chong, Sandra McCutcheon, David Parnham, Samantha Eaton, Calum MacKenzie and Fiona Houston Transmission of prion diseases by blood transfusion. J Gen Virol November 2002. — vol. 83, № 11. — р.2897-2905[недоступне посилання з липня 2019]
- Anthony E. Kincaid, Kathryn F. Hudson, Matthew W. Richey and Jason C. Bartz Rapid Transepithelial Transport of Prions following Inhalation. J. Virol. December 2012 vol. 86 no. 23 12731-12740
- Collins SJ, Lawson VA, Masters CL. Transmissible spongiform encephalopathies. Lancet.Jan 2004 — vol. 363,issue№ 9204. — p. 51-61.
- Jeffrey M, Goodbrand IA, Goodsir CM Pathology of the transmissible spongiform encephalopathies with special emphasis on ultrastructure. Micron. 1995. — vol.26, issue№ 3. — p. 277-98.
- Barron RM, Campbell SL, King D and others High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPSc in vivo. The Journal of Biological Chemistry, December 2007. — vol. 282, issue№ 49. — p. 35878-86.
- Jiri G. Safar, Holger Wille, Michael D. Geschwind, Camille Deering, Diane Latawiec, Ana Serban, David J. King, Giuseppe Legname, Karl H. Weisgraber, Robert W. Mahley, Bruce L. Miller, Stephen J. DeArmond, and Stanley B. Prusiner Human prions and plasma lipoproteins. PNAS, July 25, 2006, vol. 103 no. 30
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci, 2001. vol. 24. — p. 519-50.
- Richard Rubenstein, Binggong Chang, Perry Gray, Martin Piltch, Marie S. Bulgin, Sharon Sorensen-Melson and Michael W. Miller A novel method for preclinical detection of PrPSc in blood. Gen Virol July 2010, vol. 91 no. 7, 1883-1892 [недоступне посилання з липня 2019]
- I. P. Lipscomb, R. Herve´ , K. Harris, H. Pinchin, R. Collin and C. W. Keevil Amyloid-specific fluorophores for the rapid, sensitive in situ detection of prion contamination on surgical instruments. J Gen Virol September 2007, vol. 88, no. 9, р. 2619-2626 [недоступне посилання з липня 2019]
Див. також
Джерела
- Інфекційні хвороби (підручник) (за ред. О. А. Голубовської). — Київ: ВСВ «Медицина» (2 видання, доповнене і перероблене). — 2018. — 688 С. + 12 с. кольор. вкл. (О. А. Голубовська, М. А. Андрейчин, А. В. Шкурба та ін.) ISBN 978-617-505-675-2
- Пріонні інфекційні хвороби / К. Л. Сервецький, Є. В. Нікітін, Т. В. Чабан // Сучасні інфекції. − 2006. — № 3/4. — C.43-52.
- Семенов В.М. Клинические аспекты прионных болезней. Клиническая инфектология и паразитология (международный научно-практический журнал). 2017, том 6, № 1. с. 8-21. (рос.)
- Stanley B. Prusiner Neurodegenerative Diseases and Prions. N Engl J Med 2001; 344:1516-1526May 17, 2001DOI: 10.1056/NEJM200105173442006 (англ.)
- Herbert Budka Neuropathology of prion diseases. Br Med Bull (2003), 66 (1):121-130. doi: 10.1093/bmb/66.1.121 (англ.)
- Казаков В. Н. Прионные инфекции. (рос.)
- Max D. T. The Family That Couldn't Sleep: A Medical Mystery Paperback — Random House Trade Paperbacks.- 2007. — 336 стор. (англ.)