Ротаційно-вібраційна спектроскопія
Ротаційно-вібраційна спектроскопія — розділ молекулярної спектроскопії, що вивчає інфрачервоні та раманівські спектри молекул у газовій фазі. У формуванні цих спектрів зміни відбуваються водночас в коливальних та обертальних станах. Їх іноді називають ровібраційними. Коли такий перехід відбувається з випромінюванням або поглинанням фотона (електромагнітного випромінювання), частота фотона пропорційна різниці енергій рівнів. Фотони можна детектувати одним із спектроскопічних методів. Оскільки енергія рівнів обертання зазвичай набагато менша ніж зміна енергії коливань, зміни в обертальному стані дають так звану тонку структуру коливального спектру. Для заданого коливального переходу, ті ж теоретичні розрахунки, що й для чисто ротаційної спектроскопії дають квантові числа, рівні енергії та правила відбору. У лінійних молекулах та сферичних ротаторах, обертальні лінії знаходяться як рівномірні серії при вищих та нижчих частотах від частоти чисто коливального переходу. У молекулах типу симетричного ротатора переходи класифікують як паралельні, коли зміни дипольного моменту паралельні головній осі обертання та перпендикулярні, коли зміна перпендикулярна до цієї осі. Ровібраційний спектр молекули води, асиметричного ротатора, важливий через присутність води в атмосфері.
Загальний огляд
Ровібраційна спектроскопія має справу з молекулами в газовій фазі. Як основний, так і збуджений коливальні стнни мають послідовності квантованих обертальних рівнів. Спектри часто розділяються на лінії, що виникають внаслідок перході з одного обертального рівня в основному коливальному стані до якогось обертального рівня в збудженому коливальному стані. Лінії, що відповідають певному коливальному переходу складаються в смугу[1]. У найпростіших випадках частина інфрачервоного спектру, в якій коливальні переходи відбуваються без зміни обертального квантового числа, називається Q-гілкою. Вище за частою від Q-гілки до енергії коливального переходу додається енергія орбітального переходу. Для ΔJ = +1 це називають R-гілкою. P-гілка, для якої ΔJ = −1, має меншу частоту від Q-гілки. R-гілка виглядає дуже схоже на чисто обертальний спектр, а P-гілка виглядає майже як дзеркальне відображення R-гілки[2]. Поява обертальної тонкої структури визначається симетрією молекулярного ротатора, які класифікують аналогічно до класифікації в ротаційній спектроскопії: на лінійні молекули, сферичні, симетричні та асиметричні ротатори. Квантовомеханічна інтерпретація тонкої структури та ж, що й для чистих обертань.
За загальною конвенцією величини, що стосуються основного коливального стану, позначають подвійними штрихами, а величини, що стосуються збудженого коливального стану, - одинарними. Наприклад, обертова стала основного стану записується а аналогічна їй для збудженого - Крім того, ці сталі виражають в популярних серед молекулярних спектроскопістів одиницях см−1. тож відповідає в означенні обертової сталої для жорсткого ротатора.




Метод комбінованих різниць
Чисельний аналіз ровібраційних спектрів здається, на перший погляд, складним, оскільки частоти кожного переходу залежать від двох обертових сталих та . Однак, існують комбінації, що залежать лише від однієї обертової сталої. Для цього потрібно відняти частоти пари ліній (однієї з P-гілки, та однієї з R-гілки), які мають або однаковий нижній коливальний рівень або верхній.[3][4]. Наприклад, для двоатомної молекули лінія позначена P(J + 1) відповідає переходу (v = 0, J + 1) → (v = 1, J), а лінія R(J − 1) відповідає переходу (v = 0, J − 1) → (v = 1, J). Різниця між двома частотами відповідає різниці енергії між (J + 1) and (J − 1) рівнями нижнього коливального стану і позначається , оскільки вона є різницею між рівнями, що відрізняються тальки на дві одниці J. Коли враховують відцентрове спотворення, воно задається формулою[5]




Обертову сталу основного коливального стану B′′ та сталу відцентрового видовження D′′ можна знайти методом найменших квадратів з апроксимації цієї різниці як функції J. Сталу B′′ в чисто ротаційній спектроскопії використовують для знаходження відстані між остовами атомів в основному стані (Дивіться Додаток).
Аналогічно різниця R(J) − P(J) для збудженого коливального стану (v = 1) залежить лише від сталих B′ та D′ , і B′ можна використати для визначення міжотомних відстаней у цьому стані (чого зробити за допомогою чисто ротаційної спектроскопії неможливо).

Лінійні молекули
Двоатомні молекули з різними атомами

Двоатомні молекули з загальною формулою AB мають одну нормальну моду коливань розтягу-стиснення A-B зв'язку. Значення коливального терму [7] для ангармонічного осцилятора у першому наближенні задаються формулою


де v - коливальне квантове число , ωe частота гармонічного осцилятора, а χe - стала ангармонічності.
Коли молекула перебуває в газовій фазі, вона може обертатися навколо осі, перпендикулярної до молекулярної осі, що проходить через центр маси молекули. Обертальна енергія теж квантується, значення терму в першому наближенні задається формулою

де J - обертальне квантове число, а D - стала відцентрового спотворення. Обертова стала Bv залежить від моменту інерції молекули Iv, який змінюється для кожного коливального квантового числа v

де mA та mB — маси атомів A та B, а d — відстань між атомами. Значення термів ровібранційних станів знаходять (у наближенні Борна-Оппенгеймера), комбінуючи вирази для коливань та обертань.

Перші два члени в цьому виразі відповідають гармонічному осцилятору та жорсткому ротатору, друга пара членів вносить поправки, враховуючи ангармонічність та відцентрову дисторсію. Загальніший вираз навів Дангем.
Правила відбору, що дозволяють ровібраційні переходи, у випадку діамагнітної двоатомної молекули записуються


Переходи з Δv=±1 називають фундаментальними. Правила відбору мають два наслідки:
- Повинні змінитися обидва (коливальне та обертальне) квантові числа. перехід (Q-гілка) заборонений.
- Зміна енергії обертання може як відніматися так і додаватися до зміни енергії коливання, що призводить до утворення P- and R- гілок спектру, відповідно.

Обчислення частот переходів складніше, ніж для чистих обертань, оскільки обертова стала Bν має різне значення в основному та збудженому коливальному станах. Спрощений вираз для частот отримують, коли сталі відцентрових дисторсій та приблизно однакові[9].



де додатні m відповідають R-гілці, а від'ємні — P-гілці.
Член ω0 вказує на положення (відсутньої) Q-гілки, член означає послідовність рівновіддалених ліній у P- та R- гілках, а третій член, , відповідає за те, як відстань між сусідніми лініями змінюється зі зміною обертального квантового числа. Коли більша, ніж </math>, як це звичайно буває, проміжок між лініями зменшується зі збільшенням J у R-гілці й збільшується в P-гілці. Аналіз даних інфрачервоного спектру моноксиду карбону (чадного газу) дає значення 1.915 см−1 та 1.898 см−1. Із цих сталих легко отримати довжини зв'язків r0 = 113.3 пм, r1 = 113.6 пм[11]. Ці довжини дещо відрізняються від рівноважних. Це пояснюється нульовими коливаннями в основному коливальному стані, тоді як рівноважна довжина зв'язку обчислюється в точці мінімуму потенціальної кривої міжатомної взаємодії. Співвідношення між обертовими сталими дається формулою



де ν є коливальним квантовим числом, а α - сталою вібраційно-ротаційної взаємодії, яку можна вирахувати, коли можна знайти значення B для двох різних коливальних станів. Для моноксиду карбону req = 113.0 пм[12].
Моноксид азоту NO є особливою молекулою, оскільки вона парамагнітна, з одним неспареним електроном. Зв'язування між спіном електрона та кутовим моментом обертання призводить до лямбда-подвоєння[13] з розрахованими гармонічними частотами 1904.03 та 1903.68 см−1. Обертальні рівні розщеплюються також[14].
Двоатомні молекули з однаковими атомами
Квантова механіка для двоатомних молекул на зразок динітрогену N2 та фтор F2 якісно така ж, як для двоатомних молекул із різними атомами, але правила відбору для переходів інші. Оскільки дипольний момент цих молекул дорівнює нулю, фундаментальні коливальні переходи заборонені в дипольному наближенні. Однак, для молекули N2 слабкий квадрупольно дозволений спектр спостерігається, якщо використати довгі довжини шляхів світла як у лабораторії так і в атмосфері[15]. Спектри цих молекул можна, втім, спостерігати за допомогою раман-спектроскопії, бо вони там дозволені.
Молекула кисню є особливим випадком, оскільки вона парамагнітна, тож для дозволені магнітні дипольні переходи і їх можна спостерігати в інфрачервоному діапазоні[15]. Електронний спін, що дорівнює 1, має три можливі орієнтації щодо кутового моменту обертання N[16], тож кожен обертальний рівень розщеплений на три стани з повним кутовим моментом (обертання та спіну) , J = N + 1, N та N - 1. Кожен J стан цього так званого триплету p-типу виникає завдяки різній орієнтації спіну відносно обертання молекули[17].

Правила відбору для магнітних дипольних переходів дозволяють переходи між послідовними члена триплету (ΔJ = ±1), тож для кожного значення квантового числа обертального моменту існує два дозволених переходи. Ядро 16O має нульовий ядерний кутовий момент, тож міркування симетрії вимагають, щоб N пробігало тільки непарні значення[18][19].
Раман-спектри двоатомних молекул
Правило відбору читається

тож спектр має O-гілку (∆J = −2), a Q-гілку (∆J = 0) та S-гілку (∆J=+2). В наближенні B′′ = B′ = B частоти задаються формулами


оскільки S-гілка починається з J=0, а O-гілка з J=2. Тому, в першому наближенні проміжок між S(0) та O(2) дорівнює 12B, а відстань між сусідніми лініями дорівнює 4B як в O-, так і в S-гілках. Найбільш очевидний ефект нерівності обертових сталих B′′ ≠ B′ у тому, що Q-гілка має серію щільно розташованих сателітів завдяки переходам з ΔJ=0 для J=1,2 тощо[20]. Корисні різницеві формули, в яких знехтувано відцентровою дисторсією[21] суть:


Молекулярний кисень є особливим, бо його молекула парамагнітна, вона має два неспарені електрони[22].
Для двоатомних молекул з однаковими атомами, вагові множники ядерної спінової статистики зумовлюють змінні інтенсивності ліній між рівнями з парними та непарними . Для ядерного спіну I = 1/2 як у 1H2 та 19F2 зміна інтенсивності дорівнює 1:3. Для 2H2 та 14N2 I=1, і статистичні вагові множники дорівнюють 6 та 3, тож рівні з парними удвічі інтенсивніші. Для 16O2 (I=0) усі переходи з парними значеннями заборонені[21].

Багатоатомні лінійні молекули
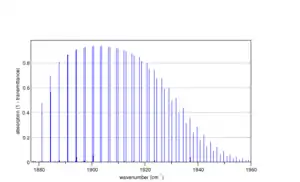
 Спектр коливань згину в 14N14N16O, розрахований програмою Spectralcalc[10]. Слабкий накладений спектр належить молекулам з ізотопом 15N, вміст якого становить 0.3% |
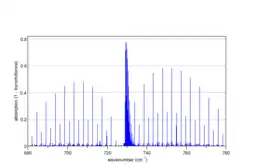 Спектр перпендикурярної моди ацетилену C2H2, розрахований програмою Spectralcalc[10], на якому видно чергування інтенсивностей 1,3 як у P- так і в R- гілках. Див. також Hollas p157 |
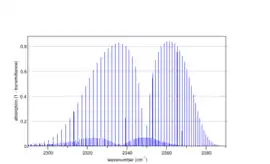 Спектр паралельної смуги асиметричної моди коливань згину в діоксиді карбону (вуглекислому газі) 12C16O2, розрахований програмою Spectralcalc[10]. Слабкий накладений спектр пояснюється поглинанням першого збудженого коливального рівня (0 11 0), який завдяки малій енергії заселений при кімнатній температурі |
Багатоатомні лінійні молекули за своєю симетрією поділяються на два класи: центросиметричні з точковою групою D∞h, сюди надежить діоксид карбону CO2 та ацетилен HCCH; і нецентросиметричні з точковою групою C∞v, такі як ціанід гідронену HCN та оксид азоту(I) NNO. Центросиметричні лінійні молекули мають нульовий дипольний момент, тож не мають чисто обертального спектру в інфрачервоному або мікрохвильовому діапазоні. З іншого боку, якщо певний коливальний стан збудженої молекули має дипольний момент, ровібраційний спектр в інфрачервоному діапазоні спостерігається.
Спектри цих молекул класифікують за напрямком вектора зміни дипольного моменту. Коли коливання створює зміну дипольного моменту вздовж молекулярної осі, спектральну смугу називають паралельною і позначають символом . Коли коливання створює зміну дипольного моменту перпендикулярно до осі молекули, смуги називають перпендикулярними і позначають символом . В обох випадках частоти P- та R- гілок підкоряються тій же тенденції, що й для двоатомних молекул. Різниця між двома класами в правилах відбору[23]. Для паралельних переходів правила відбору ті ж, що й для двоатомних молекул, тобто перехід, який відповідав би Q-гілці, заборонений. Прикладом може бути мода розтягу-скорочення зв'язку C-H у ціаніді гідрогену[24].


Для перпендикулярних коливань переходи з ΔJ=0 дозволені. Це означає, що дозволені переходи між станами з однаковим обертальним квантовим числом в основному та збудженому станах. Це стосується усіх заселених обертальних станів. Як наслідок, виникає інтенсивна, відносно широка Q-гілка, в якій лінії окремих обертальних станів перекриваються. Прикладом може бути згинальна мода в N-N-O, яку видно в області 590 см−1[10].
Спектри центросиметричних молекул мають чергування інденсивності ліній завдяки своєрідності симетрії квантових станів, оскльки поворот молекули на 180° наколо осі обертання другогго порядку аналогічне обміну місцями ідентичних ядер. У двооксиді карбону атоми Оксигену найпоширенішого ізотопу 12C16O2 мають нульовий спін і є бозонами, тож повна хвильова фунція повинна бути симетричною, коли обміняти атоми 16O місцями. Спінова частина для двох атомів зі спіном нуль завжди симетрична, тож обертова частина теж повинна бути симетричною, що справедливо лише для рівнів із парним J. Обертальні стани з непарним J не можуть існувати, і дозволені коливальні смуги складаються тільки з ліній поглинання, що відповідають парним значенням J початкового стану. Проміжок між суміжними лініями в P- та R- гілках близький до 4B, а не до 2B, оскільки лінії між ними пропали[25]. В ацетилені 1H12C12C1H два атоми гідрогену мають спін ½ і є ферміонами, тож повна хвильова функція антисиметрична, коли два ядра 1H міняються місцями. Як і в орто та пара молекулах водню ядерні спінові функції двох Гідроненів маю три симетричні орто стани й один антисиметричний пара стан. Для трьох орто станів обертальні хвильові функції повинні бути антисиметричними, що відповідає непарним J, а для одного пара стану, обертальна хвильова функція повинна бути симетричною, що відповідає парним J. Тому заселеність рівнів з непарними J втричі вища, ніж з непарними J, і інтенсивності ліній чергуються у співвідношенні 3:1[26][27].
Сферичні ротатори

Молекули, що представляються моделлю сферичного ротатора, мають однакові моменти інерції щодо довільної осі і належать до точкових груп Td (тетраедрична AX4) та Oh (октаедтична AX6). Дипольний момент таких молекул дорівнює нулю, тож вони не мають чисто ротаційних спектрів ні в інфрачервоному, ні в мікрохвильовому діапазонах[29].
Тетраедричні молекули на зразок метану CH4 мають коливання на розтяг та згин, які активні в інфрачервоному діапазоні. Вони належать до T2 (іноді вживають позначення F2) подання[30]. Ці коливання трикратно вироджені і рівні обертання мають три компоненти, розділені коріолісовою взаємодією.[31]. Значення обертальних термів задаються в першому порядку наближення формулами[32]



де є сталою коріолісової взаємодії. Правило відбору для фундаментальних коливань записується


Тож спектр дуже схожий на спектр перпендикулярних коливань лінійної молекули, з сильною Q-гілкою що в основному складається з переходів, для яких обертальне квантове число однакове в основному та збудженому станах, Ефект коріолісової взаємодії яскраво проявляється в коливаннях розтягу-скорочення C-H зв'язку метану, хоча ретельні дослідження показали, що наведена формула першого порядку наближення для коріолісової взаємодії, не зовсім годиться для метану[33][34].

Симетричні ротатори
До симетричних ротаторів належать молекули, що мають одну головну вісь обертання третього або вищих порядків симетрії. Вони мають два різні моменти інерції, а тому дві обертові сталі. Для обертання навколо будь-якої осі, перпендикулярної до головної, момент інерції , а стала обертання , як для лінійної молекули. Для обертання навколо головної осі момент інерції , а стала обертання . Серед прикладів аміак NH3 та хлорид метилу CH3Cl (обидві молекули мають симетрію, що описується точковою групою C3v), трифлуорид бору BF3 та пентахлорид фосфору PCl5 (точкова група для обох - D3h), бензол C6H6 (точкова група D6h).




Для симетричних ротаторів з повним кутовим моментом молекули асоціюють квантове число J. Для кожного значення J існує 2J+1 стан з однаковою енергією. Ці стани ідексують квантовим числом M, що приймає значення +J ...0 ... -J. Третє квантове число K квантує обертання молекули навкого головної осі. Як і для лінійних молекул переходи поділяють на паралельні () та перпендикулярні(), цього разу за напрямком зміни дипольного моменту відносно головної осі обертання. Третя категорія включає обертони та комбіновані смуги, які мають спільні риси, як з паралельними, так і перпендикулярними переходами. Чинні правила відбору
- якщо K ≠ 0, то ΔJ = 0, ±1 і ΔK = 0
- якщо K = 0, то ΔJ = ±1 і ΔK = 0
- ΔJ = 0, ±1 і ΔK = ±1
Той факт. що правила відбору різні, є підставою для виділення окремого класу і означає, що спектри виглядають по іншому, й їх часто можна відрізнити з першого погляду. Вираз для обчислення частот P- та R- гілок можна записати як[35]



де m = J+1 для R-гілки та -J для P-гілки. Три сталі відцентрової дисторсії , та потрібні для підгонки значень термів для кожного рівня[1]. Частоти дрібної структури для кожної смуги задаються як



відповідає Q-гілці дрібної структури, для якої значення частот записуються

- .

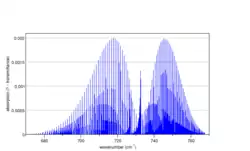 Спектр коливань на розтяг-стиснення C-Cl зв'язку в CH3Cl (паралельна смуга), розрахований програмою Spectralcalc[10] |
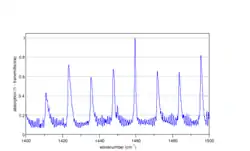 Частина спектру асиметричних коливань згину H-C-H в CH3Cl (перпендикулярна смуга), розрахована програмою Spectralcalc[10] |
Паралельні смуги
Коливання довжини зв'язку C-Cl в хлорметані CH3Cl дають паралельну смугу, оскільки зміна дипольного моменту орієнтована вздовж осі обертання третього порядку. Лінійчастий спектр має субструктуру, яку для цієї смуги видно доволі добре[10]. Насправді для того щоб повністю розділити тонку структуру потрібна спектроскопія з дуже високою роздільною здатністю. Аллен та Кросс показують частину спектру CH3D й аналізують детально експериментальні дані[36][37].
Перпендикулярні смуги
Правило відбору для перпендикулярної смуги дозволяє більше переходів, ніж для паралельної. Смугу можна розглядати як серію субструктур, кожна з яких має P, Q та R гілки. Q-гілка відділена на приблизно 2(A′-B′). Асиметричні коливання згину HCH хлорметану мають типовий вигляд. На ниж видно ряд інтенсивних Q-гілок з слабкою ротаційною тонкою структурою[10]. Аналіз спектру ускладнюється тим, що коливання основного стану зобов'язані з міркувань симетрії бути виродженими, а це означає вплив на спектр коріолісової взаємодії.[38]
Гібридні смуги
Обертони вироджених фундаментальних коливань мають складники більш, ніж одного типу симетрії. Наприклад, перший обертон коливання, що належить до E подання в молекулі на зразок аміаку NH3 мають скадові A1 та E подань. Перехід до A1 компоненти утворить паралельну смугу, а перехід до E компоненти — перпендикулярну; як наслідок утворюється гібридна смуга[39].
Інверсійні коливання в аміаку
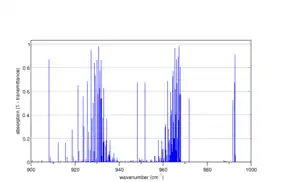 Спектр центральної частини симетричних коливань згину в аміаку, розрахований програмою[10] ілюструє подвоєння, що виникає через інверсію. |

В аміаку NH3 спостерігаються дві гілки симетричних коливань згину на частотах поблизу 930 см−1 та 965 см−1. Це так зване інверсійне подвоєння, що виникає завдяки тому, що такі коливання є фактично великоамплітудним рухом, який називають інверсією Нітрогену, В цих коливаннях атом Нітрогену проходить через площину трьох атомів Гідронену, наче вивернута парасоля. Крива потенціальної енергія має два мінімуми для двох пірамідальних геометрій, тому коливальні рівні виникають парами, які відповідають комбінаціям коливальних станів у двох мінімумах потенціалу. Комбінація двох v = 1 станів утворює симетричний стан (1+) з енергією 932.5 см−1 над основним (0+) та антисиметричний стан (1−) з 968.3 см−1[40].
Основний коливальний стан (v = 0) також роздвоєний, але різниця набагато менша, і переходи між двома рівнями можна виміряти безпосередньо в мікрохвильовому діапазоні. Вони мають частоту поблизу 24 ГГц (0.8 см−1)[41][42]. Цей перехід має історичне значення, оскільки його використали мазері на аміаку, що передував лазеру[43].
Асиметричні ротатори
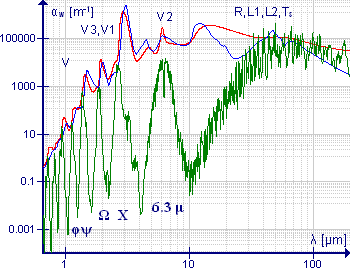
Асиметричні молекули не мають осей обертання порядку, вищого від другого. У них є три неоднакові моменти інерції відносно трьох взаємно перпендикулярних головних осей. Їхні спектри дуже складні. Частоти переходів не можна виразити аналітичними формулами, але їх можна розрахувати чисельно.
Важливим представником цього класу молекул є молекула води, оскільки водяна пара має значну концентрацію в атмосфері. Спектр низької роздільної здатності, показаний на рисунку, демонструє складність. При довжині хвилі понад 10 μ (або частотах, менших від 1000 см−1) поглинання чисто обертальне. Смуга навколо 6.3 μ (1590 см−1) виникає через коливання згину HOH; значна її ширина зумовлена обертальною тонкою структурою. Спектр високої роздільності цієї смуги наведено в книзі Аллена та Кросса на стр. 221[51]. Симетричні й асиметричні коливання розтягу-стиснення близькі між собою, тому обертальна тонка структура цих смуг перекривається. Смуги з коротшою довжиною хвилі є оберторанами та комбінаціями, у кожній з них є обертальна тонка структура. Спектри проміжної роздільної здатності смуг на 1600 см−1 та 3700 см−1 приведено в книзі Банвелла та Маккеша, стр. 91.
Ровібраційні спектри асиметричних ротаторів класифікують як A-, B- або C- типи для переходів, у яких дипольний момент змінюється вздовж осей від найменшого момента інерції до найбільшого[52].
Експериментальна методика
Зазвичай ровібраційні спектри вимінюють з високою роздільністю. Раніше цього досягали, використовуючи як дисперсивний елемент у спектрометрі дифракційну ґратку типу Ешель[14]. Дифракційна ґратка цього типу оптимізована на дифракцію вищих порядків[53]. Роздільна здатність інфрачервого Фур'є-спетрометра залежить від максимального запізнення сигналів від рухомих дзеркал. Наприкад, щоб досягти роздільності 0,1 см−1, рухоме дзеркало повинно переміститися на 10 см при нульовій різниці шляхів. Конн виміряв ровібраційний спектр CO2 у атмосфері Венери саме з таким розділенням[54]. Зараз комерційно доступні спектрометри з розділенням 0,001 см−1. Загальною перевагою інфрачервоних Фур'є-спетрометрів є те, що використання як дисперсивного елемента монохроматора з аналогічним розділенням означало дуже вузькі вхідну та вихідну апертури.
Вимірюючи спектри газів доволі легко отримати дуже великі шляхи пробігу для світлового променя за допомогою багаторазового відбиття у вимірювальній комірці[55]. Це важливо тому, що дозволяє зменшити тиск газу й тим самим мінізувати уширення спектральних ліній, що могло б погіршити розділення. Комерційно доступні шляхи до 20 м.
Додаток
Метод комбінованих різниць використовує різниці частот в P- та R- гілках, щоб отримати дані, які залежали б тільки від обертових сталих основного чи збудженого коливального стану. Для збудженого стану:

Цю залежність можна підігнати, використовуючи метод найменших квадратів до експериментальних даних для моноксиду карбону[56]. Дані, розраховані за формулою

де відцентровою дисторсією знехтувано, показані в стовпчику таблиці з індексом (1). Формула означає, що дані повинні лежати на прямій з нахилом 2B′′, яка б проходила через нуль. На перший погляд, дані наче узгоджуються з моделлю з середньо-квадратичним розкидом 0,21 см−1. Однак, коли врахувати відцентрову дисторсію за формулою : апроксимація за методом найменших квадратів значно покращується, а розкид зменшується до 0,000086 см−1. Розраховані дані наведено в стовпчику (2).

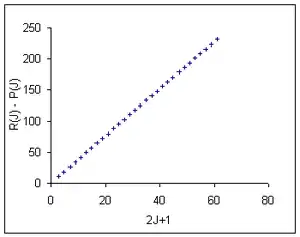
| J | 2J+1 | Розрахунок (1) | Залишок (1) | Розрахунок (2) | Залишок (2) | |
|---|---|---|---|---|---|---|
| 1 | 3 | 11.4298 | 11.3877 | 0.0421 | 11.4298 | −0.000040 |
| 2 | 5 | 19.0493 | 18.9795 | 0.0698 | 19.0492 | 0.000055 |
| 3 | 7 | 26.6680 | 26.5713 | 0.0967 | 26.6679 | 0.000083 |
| 4 | 9 | 34.2856 | 34.1631 | 0.1225 | 34.2856 | 0.000037 |
| 5 | 11 | 41.9020 | 41.7549 | 0.1471 | 41.9019 | 0.000111 |
| 6 | 13 | 49.5167 | 49.3467 | 0.1700 | 49.5166 | 0.000097 |
| 7 | 15 | 57.1295 | 56.9385 | 0.1910 | 57.1294 | 0.000089 |
| 8 | 17 | 64.7401 | 64.5303 | 0.2098 | 64.7400 | 0.000081 |
| 9 | 19 | 72.3482 | 72.1221 | 0.2261 | 72.3481 | 0.000064 |
| 10 | 21 | 79.9536 | 79.7139 | 0.2397 | 79.9535 | 0.000133 |
| 11 | 23 | 87.5558 | 87.3057 | 0.2501 | 87.5557 | 0.000080 |
| 12 | 25 | 95.1547 | 94.8975 | 0.2572 | 95.1546 | 0.000100 |
| 13 | 27 | 102.7498 | 102.4893 | 0.2605 | 102.7498 | −0.000016 |
| 14 | 29 | 110.3411 | 110.0811 | 0.2600 | 110.3411 | 0.000026 |
| 15 | 31 | 117.9280 | 117.6729 | 0.2551 | 117.9281 | −0.000080 |
| 16 | 33 | 125.5105 | 125.2647 | 0.2458 | 125.5105 | −0.000041 |
| 17 | 35 | 133.0882 | 132.8565 | 0.2317 | 133.0882 | 0.000035 |
| 18 | 37 | 140.6607 | 140.4483 | 0.2124 | 140.6607 | 0.000043 |
| 19 | 39 | 148.2277 | 148.0401 | 0.1876 | 148.2277 | −0.000026 |
| 20 | 41 | 155.7890 | 155.6319 | 0.1571 | 155.7891 | −0.000077 |
| 21 | 43 | 163.3443 | 163.2237 | 0.1206 | 163.3444 | −0.000117 |
| 22 | 45 | 170.8934 | 170.8155 | 0.0779 | 170.8935 | −0.000053 |
| 23 | 47 | 178.4358 | 178.4073 | 0.0285 | 178.4359 | −0.000093 |
| 24 | 49 | 185.9713 | 185.9991 | −0.0278 | 185.9714 | −0.000142 |
| 25 | 51 | 193.4997 | 193.5909 | −0.0912 | 193.4998 | −0.000107 |
| 26 | 53 | 201.0206 | 201.1827 | −0.1621 | 201.0207 | −0.000097 |
| 27 | 55 | 208.5338 | 208.7745 | −0.2407 | 208.5338 | −0.000016 |
| 28 | 57 | 216.0389 | 216.3663 | −0.3274 | 216.0389 | 0.000028 |
| 20 | 59 | 223.5357 | 223.9581 | −0.4224 | 223.5356 | 0.000128 |
| 30 | 61 | 231.0238 | 231.5499 | −0.5261 | 231.0236 | 0.000178 |

Література
- Allen, H.C.; Cross, P.C. (1963). Molecular vib-rotors; the theory and interpretation of high resolution infra-red spectra. New York: Wiley.
- Atkins, P.W.; de Paula, J. (2006). Physical Chemistry (вид. 8th). Oxford University Press. с. 431–469. ISBN 0198700725. Chapter (Molecular Spectroscopy), Section (Vibration-rotation spectra) and page numbers may be different in different editions.
- Banwell, Colin N.; McCash, Elaine M. (1994). Fundamentals of molecular spectroscopy (вид. 4th). McGraw-Hill. с. 40. ISBN 0-07-707976-0.
- Hollas, M.J. (1996). Modern Spectroscopy (вид. 3rd). Wiley. ISBN 0471965227.
- Straughan, B.P.; Walker, S. (1976). Spectroscopy 2 (вид. 3rd). Chapman and Hall. с. 176–204. ISBN 0-470-15032-7.
Виноски
- Hollas p101
- Традиційно інфрачервоні спектри показуються з частотами, що зменшуються зліва направо, що відповідає зростанню довжини хвилі. Сучасніші тексти можуть показувати частоти зі зростанням зліва направо. P-гілка завжди має менші частоти, ніж Q-гілка.
- Hollas p132
- Atkins та de Paula p458 з діаграмами
- Allen and Cross, p 116
- Експериментальні спектри фундаментальної смуги (їх показано на рисунку) та першої обертонної смуги (у разі подвійного збудження поблизу 2 × 2140 см−1) можна подивитисяс в Banwell and McCash, p 67
- Значення терму прямо зв'язане з енергією
- Переходи з ∆v≠1 називають обертонами. Вони заборонені в гармонічному наближенні, але їх можна бачиит як слабкі смуги завдяки ангармонічності
- Allen and Cross, p113
- Simulated spectrum created using infrared gas spectra simulator
- Banwell and McCash, p 70
- Banwell and McCash, p69
- Інший приклад лямбда подвоєння спостерігається у спектрі радикала гідроксилу.
- Gillette, R. H.; Eyster,Eugene H. (1939). The Fundamental Rotation-Vibration Band of Nitric Oxide. Phys. Rev. 56 (11): 1113–1119. Bibcode:1939PhRv...56.1113G. doi:10.1103/PhysRev.56.1113.
- Goldman, A.; Reid, J.; Rothman, L. S. (1981). Identification of electric quadrupole O2 and N2 lines in the infrared atmospheric absorption spectrum due to the vibration‐rotation fundamentals. Geophysical Research Letters 8 (1): 77. Bibcode:1981GeoRL...8...77G. doi:10.1029/GL008i001p00077.
- Деякі тексти позначають це квантове число літерою K
- Hollas p116
- Strandberg, M. W. P.; Meng, C. Y.; Ingersoll, J. G. (1949). The Microwave Absorption Spectrum of Oxygen. Phys.Rev. 75 (10): 1524–1528. Bibcode:1949PhRv...75.1524S. doi:10.1103/PhysRev.75.1524.pdf
- Krupenie, Paul H. (1972). The Spectrum of Molecular Oxygen. J. Phys. Chem. Ref. Data 1, 423 (1972) 1 (2): 423–534. Bibcode:1972JPCRD...1..423K. doi:10.1063/1.3253101.
- Hollas, pp 133–135. Стоксова сторона спектру моноксиду карбону показана на p134
- Hollas, p135
- Fletcher, William H.; Rayside, John S. (1974). High resolution vibrational Raman spectrum of oxygen. Journal of Raman Spectroscopy 2 (1): 3–14. Bibcode:1974JRSp....2....3F. doi:10.1002/jrs.1250020102.
- Straughan and Walker, vol2, p185
- The spectrum of this vibration mode, centered at ca. 3310 cm−1 is shown in Banwell and McCash, p76, and also in Hollas, p156
- Straughan and Walker, vol2 p186
- Hollas p155
- Straughan and Walker vol2, pp 186−8
- PGOPHER a Program for Simulating Rotational Structure, C. M. Western, University of Bristol
- Слабенький спектр спостерігається завдяки полярності збуджених станів. Детальніше в статі ротаційна спектроскопія.
- Термін подання вживається в теорії груп для класифікації дії операцій симетрії на, в цьому випадку, коливання в молекулі.
- Straughan and Walker vol2, p199
- Allen and Cross, p67
- Jahn, H.A. (1938). Proceedings of the Royal Society A168: 469, 495.; A171. 1939. с. 450. Пропущений або порожній
|title=(довідка) - Hecht, K.T. (1960). Vibration-rotation energies of tetrahedral XY4 molecules: Part II. The fundamental ν3 of CH4. J. Mol. Spectrosc. 5: 335, 390. Bibcode:1961JMoSp...5..390H. doi:10.1016/0022-2852(61)90103-5.
- Allen and Cross, p131
- Allen and Cross pp 134–148
- Allen, H.C. Jr.; E.K.Pyler (1959). Some Vibrational-Rotational Bands of Deuterated Methanes. J. Research Nat. Bur. Standards 63A (2): 145–153. doi:10.6028/jres.063a.007.
- Allen and Cross, pp 149–164 has a detailed analysis.
- Allen and Cross, pp 164–70.
- Hollas p167 дає 0.79 см−1 для різниці 0+−0−, 931.7 см−1 для 0−−1+ і 35.8 см−1 для 1+−1−.
- Harris, Daniel, C.; Bertolucci, Michael, D. (1978). Symmetry and Spectroscopy. OUP. с. 168–170. ISBN 0-19-502001-4.
- Spectra are shown in Allen and Cross, pp 172–174
- Straughan and Walker p124
- Bertie J. E.; Lan Z. (1996). Infrared Intensities of Liquids XX: The Intensity of the OH Stretching Band of Liquid Water Revisited, and the Best Current Values of the Optical Constants of H2O(l) at 25°C between 15,000 and 1 cm−1. Applied Spectroscopy 50 (8): 1047–1057. Bibcode:1996ApSpe..50.1047B. doi:10.1366/0003702963905385. Процитовано 8 серпня 2012.
- Spectroscopy of Atmospheric Gases (spectral databases). V.E. Zuev Institute of Atmospheric Optics SB RAS. Архів оригіналу за квітень 16, 2013. Процитовано 8 серпня 2012. «... various data sources: HITRAN and GEISA spectral databanks, original data obtained by IAO researchers in collaboration with other scientists, H2O spectra simulated by Partridge and Schwenke etc... ...»
- Aringer B.; Kerschbaum F.; Jørgensen U. G. (2002). H2O in stellar atmospheres. Astronomy and Astrophysics (EDP Sciences) 395: 915–927. Bibcode:2002A&A...395..915A. doi:10.1051/0004-6361:20021313. Процитовано 8 серпня 2012.
- Warren S. G. (1984). Optical constants of ice from the ultraviolet to the microwave. Applied Optics 23: 1206. Bibcode:1984ApOpt..23.1206W. doi:10.1364/AO.23.001206. Процитовано 8 серпня 2012.
- Warren S. G.; Brandt R. E. (2008). Optical constants of ice from the ultraviolet to the microwave: A revised compilation. J. Geophys. Res. 113. Bibcode:2008JGRD..11314220W. doi:10.1029/2007JD009744. Процитовано 8 серпня 2012.
- Wozniak B.; Dera J. (2007). Atmospheric and Oceanographic Sciences Library. New York: Springer Science+Business Media. LLC. ISBN 978-0-387-30753-4. Процитовано August 4, 2012.
- Hitran on the Web Information System. Harvard-Smithsonian Center for Astrophysics (CFA), Cambridge, MA, USA; V.E. Zuev Institute of Atmosperic Optics (IAO), Tomsk, Russia. Процитовано 11 серпня 2012.
- Dalby, F.W.; Nielsen, H.H. (1956). Infrared Spectrum of Water Vapor. Part I—The 6.26μ Region. J. Chem. Phys. 25 (5): 934–940. Bibcode:1956JChPh..25..934D. doi:10.1063/1.1743146.
- Allen and Cross, chapter 8.
- Hollas, pp. 38−41. Приклад 3.1 показує як роздільна здатність співвідноситься з дифракційним порядком та відстанню між лініями в ґратці
- Connes, J.; Connes, P. (1966). Near-Infrared Planetary Spectra by Fourier Spectroscopy. I. Instruments and Results. Journal of the Optical Society of America 56 (7): 896–910. doi:10.1364/JOSA.56.000896.
- Patricia B. Coleman, ред. (1993). Practical Sampling Techniques for Infrared Analysis. CRC PressINC. ISBN 0-8493-4203-1.
- Harris, Daniel, C.; Bertolucci, Michael, D. (1978). Symmetry and Spectroscopy. OUP. с. 125. ISBN 0-19-502001-4.
