Інфрачервона спектроскопія
Інфрачерво́на спектроскопі́я, ІЧ спектроскопі́я — різновид молекулярної оптичної спектроскопії, оснований на взаємодії речовини з електромагнітним випромінюванням в ІЧ діапазоні: між червоним краєм видимого спектра (хвильове число 14000 см−1) і початком короткохвильового радіодіапазону (20 см−1).

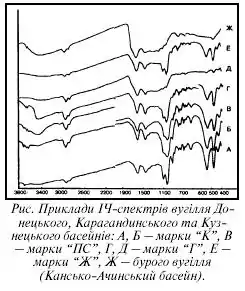
ІЧ спектри виникають при поглинанні ІЧ випромінення на частотах, що збігаються з деякими власними коливальними і обертальними частотами молекул або з частотами коливань кристалічної ґратки. ІЧ спектри отримують за допомогою спектрометрів різних типів, робочий діапазон яких знаходиться в межах так званої фундаментальної ІЧ області (400 см−1 — 4000 см−1).
Найпоширенішим способом підготовки зразків для інфрачервоної спектроскопії є пресування зразку в таблетку з KBr.
На основі ІЧ спектрів можна проводити якісний та кількісний аналіз речовини.
Інфрачервона спектроскопія дозволяє отримувати спектри речовини у всіх її агрегатних станах.
Інфрачервона спектроскопія відбивання використовується при дослідженні твердих тіл, особливо монокристалів. Для зразків із сильним поглинанням і поверхневих сполук розроблений так званий метод порушеного повного внутрішнього відбиття.
Інфрачервона спектроскопія застосовується для виявлення і оцінки фаз, вміст яких у руді та гірських породах перевищує 1-5 %. Вона — джерело інформації для вирішення таких питань кристалохімії, як будова складних комплексних аніонів, ізоморфних заміщень у мінералах тощо.
Успішно використовується ІЧ спектроскопія для вивчення флотаційних реагентів, міжфазної зони «адгезив-субстрат», ідентифікації і кількісних вимірювань промислових забруднень, аналізу в польових умовах, вивчення реакцій в атмосфері та ін.
Історія методу

Інфрачервоне випромінювання відкрив 1800 року астроном Вільям Гершель. Використовуючи призму, він спостерігав підвищення температури в області, що розташовується за червоною межею спектру видимого випромінювання. У 1882—1900 роках Вільям Ебней і Едвард Фестінг записали інфрачервоні спектри 52 сполук і зіставили спостережувані смуги поглинання з функціональними групами, наявними у цих молекулах. Суттєвий внесок у метод зробив американський фізик Вільям Кобленц, який з 1903 року, користуючись призмою з хлориду натрію, отримав досить точні та повні ІЧ-спектри для сотень органічних і неорганічних речовин[1][2].
Перші експерименти з реєстрації інфрачервоних спектрів були вкрай трудомісткими, оскільки дослідники були змушені збирати власні прилади, шліфувати та полірувати призми, сріблити дзеркала, градуювати прилади за показниками заломлення кам'яної солі. При цьому спектрометри були чутливі до вібрацій, тому їх розташовували на фундаменті, а дослідження виконували вночі. Час реєстрації одного спектра становив від 3 до 4 годин. Уже в ранніх роботах було показано, що ІЧ-спектри сполук мають індивідуальний вигляд[1].
В той час природа поглинання інфрачервоного випромінювання не була до кінця з'ясована, однак до 1930-х років була створена теорія, в якій вважалося, що це поглинання спостерігається внаслідок коливань молекул і що характер цього поглинання так чи інакше пов'язаний зі зміною дипольного моменту, правилами відбору, симетрією молекул і ступенем ангармонізму коливань[2].
1940 року фірми Dow Chemical і American Cyanamid створили власні однопроменеві прилади для вивчення вуглеводнів. Комерційні спектрометри почали випускатися 1946 року при співпраці American Cyanamid з Perkin-Elmer. Доступність приладів призвела до створення обширних таблиць кореляції спостережуваних смуг поглинання зі структурою поглинальних функціональних груп[3].
Після Другої світової війни з'явилася можливість підсилювати слабкий сигнал ІЧ-спектрометрів, що скоротило час експерименту до 1—2 годин. Потім була вдосконалена техніка виготовлення термоелектронних приймачів з малим часом відгуку. Ці покращені детектори дозволяли уникнути дрейфу показів у часі та призвели до створення двохпроменевих приладів, де шкала калібрувалася у відсотках пропускання навпроти шкали довжин хвиль чи хвильових чисел[1].
Стало можливим промислове отримання великих і якісних кристалів галогенідів лужних металів, необхідних для створення оптичних елементів приладів, що дозволило подолати багато труднощів. Наприклад, синтетичний бромід калію, на відміну від кам'яної солі, що використовувалася раніше, дозволив записувати ІЧ-спектри аж до 400 см−1, в той час як попередня межа становила 650 см−1[4].
Розквітом ІЧ-спектроскопії стала поява ІЧ-інтерферометрів, які спочатку використовувалися для детектування дуже слабкого інфрачервоного випромінювання астрономічних об'єктів. Після розробки швидких методів конвертування інтерферограм у спектри (перетворення Фур'є) та зменшення часу сканування подібні прилади почали випускатися серійно, що в 1970-ті роки дозволило вийти на ринок ІЧ-спектрометрів компаніям, які виготовляли комп'ютери, але не мали досвіду роботи в області спектроскопії (Nicolet, Bruker). Перевага ІЧ-інтерферометрів полягала в їхній мультиплексності (перевага Фелгетта), тобто одночасному зборі інформації про поглинання усіх довжин хвиль, за рахунок чого досягалося вище відношення сигналу до шуму для фіксованого часу сканування спектра. Другою перевагою була продуктивність приладів нового типу: в той час як дисперсійні прилади мали вхід і вихід, що обмежували кількість світла, що проходило крізь них, продуктивність інтерферометра визначалася товщиною пучка світла від джерела. Ймовірно, мода також відіграла значну роль у поширенні ІЧ-спектрометрів з перетворенням Фур'є, оскільки в той час не було великої необхідності у високому співвідношенні сигнал/шум: зразки зазвичай готувалися набагато довше, ніж виконувалися вимірювання, і маса зразків була достатньою для запису якісних спектрів[5].
ІЧ-інтерферометри дозволили отримувати спектри в дальній ІЧ-області, спостерігати ґраткові коливання кристалів, а також, завдяки високому відношенню сигнал/шум, долати труднощі при інтерпретації спектрів органічних сполук. Одним із популярних занять у той час була цифрова обробка спектрів, а саме вилучення смуг поглинання розчинників, визначення ступеню чистоти й характеру домішок. Інтерферометри почали широко використовуватися в дослідженні водних розчинів біологічних молекул[6].
В 1980-ті роки з'явилися комбіновані методи, що об'єднали газову хроматографію та ІЧ-спектроскопію. Підлогові прилади великого розміру змінилися компактнішими настільними моделями. З'явилася можливість ступінчастого сканування в часі, що дозволило вивчати динамічні процеси зі збором даних в одній точці[6].
Принцип методу
Головні характеристики ІЧ-випромінювання
Інфрачервона спектроскопія із хвиловими числами менше 100 см-1 при поглинанні органічною молекулою перетвоюється у енергію обертання. Поглинання є квантованим, тому обертальний спектр молекул складається із дискретних ліній.
Випромінювання у інтервалі 10 000-100 см-1 при поглинанні перетворюється органічною молекулою на енергію коливань. Це поглинання також квантоване, але коливальний спектр складається не з ліній, а із смуг, оскільки кожний коливальний перехід супроводжується змінами обертальних станів.
Поглинання смуг у спектрах позначається через хвильові числа , які вимірюють у зворотних сантиметрах см-1; ця одиниця є прямо пропорційною енергії коливання, а шкали спектрометрів є лінійними відносно см-1. Хвильові числа також називають "частотами". Це неправильно, оскільки хвильові числа у см-1 дорівнюють у мкм, а частоти у Гц дорівнюють у см, де - швидкість світла.





ІЧ-спектроскопія базується на явищі поглинання хімічними речовинами інфрачервоного випромінювання з одночасним збудженням коливань молекул. Інфрачервоне випромінювання є електромагнітною хвилею та характеризується довжиною хвилі λ, частотою ν і хвильовим числом , які пов'язані наступною залежністю:


де c — швидкість світла, а n — показник заломлення середовища[7].
У спектроскопії поглинання, частковим випадком якої є ІЧ-спектроскопія, відбувається поглинання молекулами фотонів певної енергії, яка пов'язана з частотою електромагнітної хвилі через сталу Планка:

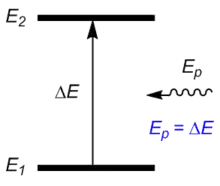
При поглинанні фотона відбувається збудження — збільшення енергії молекули: вона переходить із основного коливального стану E1 в деякий збуджений коливальний стан E2 так, що енергетична різниця між цими рівнями дорівнює енергії фотона[7].

Енергія поглинутого інфрачервоного випромінювання витрачається на збудження коливальних переходів для речовин у конденсованому стані. Для газів поглинання кванта ІЧ-випромінювання викликає коливальні й обертальні переходи[7].
Види та енергія коливань молекул
Коливальні рухи молекул визначаються їхніми внутрішніми, або коливальними, ступенями вільності. Кількість коливальних ступенів вільності та відповідних їм нормальних[K 1] коливань дорівнює (3n – 5) для лінійних молекул і (3n – 6) для нелінійних молекул, де n — кількість атомів у молекулі[K 2]. Наприклад, молекула води H2O нелінійна та має 3 коливальних ступені вільності, а лінійна молекула водню H2 — лише одну[8][9].
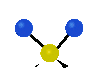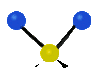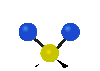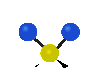
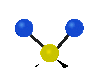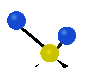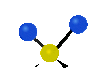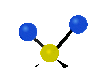
Коливання молекул можуть полягати у зміні довжин зв'язків (валентні коливання, v) або кутів між зв'язками (деформаційні коливання, δ). Валентні коливання можуть бути симетричними та антисиметричними, а деформаційні коливання поділяються на ножничні, маятникові, віялові та крутильні. Для складніших молекул, в яких одна з частин, що коливається деформаційно, є набагато масивнішою за іншу, деформаційні коливання частіше описують як площинні та позаплощинні. Коливання, які полягають в одночасній зміні кількох довжин зв'язків або валентних кутів, називаються скелетними[10].
| Валентні коливання (stretching) | Деформаційні коливання | ||||
|---|---|---|---|---|---|
| симетричне | антисиметричне | площинні | позаплощинні | ||
| ножничне
(scissoring) |
маятникове
(rocking) |
віялове
(wagging) |
крутильне
(twisting) | ||
 |
 |
 |
 |
 |
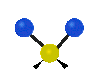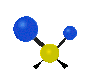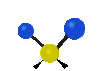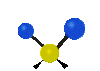 |
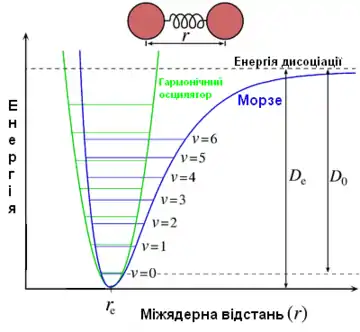
Коливання молекул можуть бути описані з допомогою моделей гармонічного й ангармонічного осцилятора. З точки зору моделі гармонічного осцилятора, двохатомна молекула є двома масами m1 і m2, з'єднаними пружною пружиною, що не має маси, з силовою сталою K. В такому випадку частота коливань атомів такої молекули вздовж лінії, що проходить через центри їхніх мас, дорівнює[11]:


З цих виразів випливає, що спостережувана частота коливань двохатомного осцилятора залежить від силової сталої K, яка, в свою чергу, пов'язана з енергією зв'язку між двома атомами, а також від маси атомів, які беруть участь в коливанні. Для багатоатомних молекул коливання мають складніший характер і наближення гармонічного осцилятора не застосовне[11].
Використовуючи закон Гука, можна зробити приблизне віднесення частот валентних коливань. Два атома і зв'язок між ними розглядаються як гармонічний осцилятор, який складається із двох мас, сполучених пружинкою. Частота коливання із масами атомів пов'язані із силовою сталою

де - хвильове число (см-1), - швидкість світла (см/c), - силова стала зв'язку (дин/см), - відповідно маси атомів та (г).[12]




Потенціальна енергія гармонічного осцилятора пов'язана з відхиленням відстані між атомами X наступним чином[11]:

Графік потенціальної енергії є параболою, симетричною відносно початкового положення атомів у стані спокою (re). Згідно з квантовою механікою, енергетичні стани молекули квантовані, тобто є дискретними. Подібні квантовані стани називають коливальними рівнями. Коливальні рівні розташовані один від одного на однаковій відстані, і їхню енергію можна обчислити з рівняння[11]

При vi = 0 молекула перебуває на найнижчому коливальному рівні, і коливальна енергія в такому стані дорівнює E = ½ hν. Молекула завжди має цю енергію, її не можна відібрати. В наближенні гармонічного осцилятора дозволені лише переходи з Δv = ±1, тобто лише на сусідні рівні (правило відбору)[11].
Більш точною є модель ангармонічного осцилятора. Ангармонічність проявляється, якщо величина дипольного моменту змінюється не пропорційно до зміщення атомів. Відмінність цієї моделі полягає в тому, що відстань між коливальними рівнями зменшується зі збільшенням номера рівня. Відхилення від гармонічності також збільшується знизу вгору. Енергія рівня у випадку ангармонічного осцилятора виражається як[11]:

Ангармонічність коливань призводить до послаблення строгості правила відбору, внаслідок чого у спектрах можуть спостерігатися переходи з Δv = ±2 — обертони. Зазвичай, частота обертону потрапляє в область 2×ν1-b, де b = 2—10 см−1. Можливим є також виникнення комбінаційних, або складених, смуг, що мають частоту ν1 + ν2, де ν1 і ν2 — частоти якихось фундаментальних коливань молекули. Комбінаційна смуга з'являється при коливальних переходах зі збуджених станів. Зазвичай для конденсованого стану інтенсивність обертонів і комбінаційних смуг у 10—100 разів нижча, ніж основних, хоча можуть зустрічатися й винятки[13].
Якщо обертон або комбінаційна смуга збігаються за частотою з якимось фундаментальним коливанням, проявляється резонанс Фермі, який призводить до появи двох смуг поглинання приблизно однакової інтенсивності, в той час як очікується наявність лише однієї фундаментальної смуги. Іноді також відбувається змішування коливань з приблизно однаковою частотою: при цьому кількість коливань залишається такою ж, але вони проявляються при інших частотах і вже не можуть бути віднесені лише до одного зв'язку. Ускладнюючим фактором також є поява у спектрах тонкої структури, що відповідає обертальним переходам (таке явище спостерігається лише для речовин у газоподібному стані)[10].
Характеристичні коливання
Багатоатомні молекули мають 3n – 6(5) нормальних коливань, і в кожному такому коливанні беруть участь не пари атомів при одному зв'язку, а в тій чи іншій мірі всі n атомів молекули. Однак було експериментально встановлено, що для коливань деяких функціональних груп внесок «сторонніх» атомів і зв'язків достатньо малий, тому незалежно від оточення ці функціональні групи поглинають в обмеженому інтервалі частот. Цей факт дозволив шляхом порівняння багатьох спектрів співвіднести наявність у молекулі характерних фрагментів зі спостережуваними смугами поглинання. Такі смуги отримали назву групових, або характеристичних. За ними можна швидко й однозначно підтвердити наявність або відсутність у молекулі відповідних фрагментів[14].
Виникнення характеристичних коливань може відбуватися з двох причин[15]:
- Якщо характеристичне коливання стосується легкого атома, зв'язаного з важким, то практично увесь рух зосереджено саме на ньому, і вплив решти молекули на нього досить слабкий.
- Коливання, що стосуються атомів дуже близької маси (наприклад, C=O, C≡N), слабко взаємодіють з коливаннями інших частин молекули.
Існують також менш визначені характеристичні коливання, які спостерігаються у порівняно ширшому інтервалі частот. Однак їхнє розташування у спектрі можна пояснити масою атомів, резонансом або електронними ефектами в молекулі[15].
Поглинання випромінювання
Зазвичай в експерименті прилад випромінює одночасно всі довжини хвиль інфрачервоного випромінювання, включаючи ближню ІЧ-область (14 000—4000 см−1), середню ІЧ-область (4000—400 см−1) та дальню ІЧ-область (400—10 см−1). Поглинання випромінювання речовиною кількісно описується законом Бугера — Ламберта — Бера, а спектр отримується при побудові залежності пропускання (T, англ. transmittance, %) чи оптичної густини (D, англ. optical density) від довжини хвилі (частоти, хвильового числа)[16].
Для того, щоб відбулося поглинання випромінювання, необхідне виконання двох умов. По-перше, поглинаються лише хвилі такої частоти, яка дорівнює частоті того чи іншого коливання молекули. По-друге, коливання повинно викликати зміну дипольного моменту молекули. Через це молекули, що не мають дипольного моменту (наприклад, H2, N2, O2, а також солі без ковалентних зв'язків і метали), не поглинають інфрачервоне випромінювання. Інтенсивність смуг в ІЧ-спектрі пропорційна до квадрата зміни дипольного моменту[16][17].
Див. також
Коментарі
- Нормальними називаються коливання, які не залежать від інших коливань.
- Цей вираз отримується з того факту, що кожен із n атомів молекули має 3 ступеня вільності в тривимірному просторі, а отже сумарно молекула має 3n ступенів вільності. З них три — це поступальні ступені вільності, пов'язані з переміщенням цілої молекули у просторі, ще дві чи три — обертальні ступені вільності (для лінійної молекули одна зі ступенів вільності вироджується, оскільки не призводить до зміни енергії). Відповідно, інші ступені вільності є коливальними.
Примітки
- Смит, 1982, с. 9—11.
- ESS, 2010, с. 2938.
- Larkin, 2011, с. 4.
- ESS, 2010, с. 2941.
- ESS, 2010, с. 2943—2944.
- ESS, 2010, с. 2944.
- Larkin, 2011, с. 7—8.
- Larkin, 2011, с. 8—9.
- Смит, 1982, с. 145.
- Stuart, 2004, с. 8—13.
- Larkin, 2011, с. 10—13.
- Robert M.Silverstein, Francis X. Webster, David J. Kiemle - Spectrometric identification of organic compounds.
- Смит, 1982, с. 151—152.
- Смит, 1982, с. 153.
- Смит, 1982, с. 153—154.
- Larkin, 2011, с. 13—15.
- Бёккер, 2009, с. 141.
Література
- Мала гірнича енциклопедія : у 3 т. / за ред. В. С. Білецького. — Д. : Донбас, 2004. — Т. 1 : А — К. — 640 с. — ISBN 966-7804-14-3.
- Глосарій термінів з хімії / уклад. Й. Опейда, О. Швайка ; Ін-т фізико-органічної хімії та вуглехімії ім. Л. М. Литвиненка НАН України, Донецький національний університет. — Дон. : Вебер, 2008. — 738 с. — ISBN 978-966-335-206-0.
- Бёккер Ю. Спектроскопия = Spektroskopie / Пер. с нем. Л. Н. Казанцевой, под ред. А. А. Пупышева, М. В. Поляковой. — М. : Техносфера, 2009. — 528 с. — ISBN 978-5-94836-220-5. (рос.)
- Смит А. Прикладная ИК-спектроскопия: основы, техника, аналитическое применение / Пер. с англ. Б. Н. Тарасевича, под ред. А. А. Мальцева. — М. : Мир, 1982. — 328 с. (рос.)
- Encyclopedia of Spectroscopy and Spectrometry / Lindon J. — 2nd Ed. — Academic Press, 2010. — 3312 p. (англ.)
- Larkin P. J. Infrared and raman spectroscopy: principles and spectral interpretation. — Elsevier, 2011. — 230 p. — ISBN 978-0-12-386984-5. (англ.)
- Stuart B. H. Infrared Spectroscopy: Fundamentals and Applications. — Wiley, 2004. — 242 p. (англ.)