Ротаційна спектроскопія
Ротаційна спектроскопія має справу з вимірюваннями енергії переходів між квантованими обертальними станами молекул в газовій фазі. Спектри полярних молекул можна виміряти через поглинання або випромінювання в мікрохвильовому діапазоні[2] або за допомогою інфрачервоної спектроскопії. Обертальні спектри неполярних молекул так виміряти не можна, але їх можна отримати за допомогою раманівської спектроскопії. Обертальні спектри іноді називають чистими, щоб відрізнити від ротаційно-вібраційної спектроскопії або навіть вібронної спектроскопії, коли обертальні, коливальні та електронні переходи відбуваються водночас.
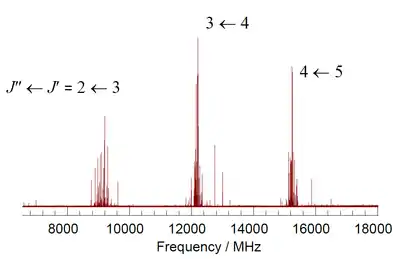
З огляду на ротаційну спектроскопію молекули класифікують за симетрією як сферичні ротатори, лінійні ротатори та симетричні ротатори, для яких можна отримати аналітичні вирази для енергії обертань. Для асиметричних ротаторів аналітичні вирази існують до J=3, але розрахунок вищих рівнів вимагає чисельних методів. Теоретичні значення енергії отримують, розглядаючи молекули як жорсткі ротатори, а потім враховуючи додаткові члени, пов'язані з центрифугаційними спотворення, тонку структуру, надтонку структуру та сили Коріоліса. Співставлення експериментальних спектрів з дає чисельні значення для моментів інерції, з яких можна в окремих випадках отримати дуже точні значення довжини хімічних зв'язків та кутів. В електростатичному полі проявляється ефект Штарка, що дозволяє визначити електричний дипольний момент.
Важливим застосуванням ротаційної спектроскопії є дослідження хімічного складу міжзоряного простору за допомогою радіотелескопії.
Застосування
Ротаційну спектроскопію використовують у першу чергу для дослідження фундаментальних аспектів молекулярної фізики. Вона є унікальним точним інструментом визначення структури молекул у газовій фазі. Її можна використати для встановлення бар'єрів, зв'язаних з обертанням групи CH3I відносно інших груп у хлоротолуені[3]. Коли можна спостерігати тонку або надтонку структуру, метод може дати інформацію про електронну структуру молекули. За допомогою ротаційної спектроскопії встановлено значну частину сучасних уявлень про слабку взаємодію на зразок Ван дер Ваальсової, водневого та гологенного зв'язків. У радіоастрономії метод має вирішальну роль у з'ясуванні хімічного складу міжзоряного середовища. Мікрохвильові переходи вимірюють в лабораторії й порівнюють з космічними випромінюваннями. Першою стабільною багатоатомною молекулою, яку виявили в міжзоряному просторі була молекула аміаку[4]. Вимірювання моноксиду хлору[5] важливе для хімії атмосфери. Нові астрохімічні проекти використовують як лабораторні вимірювання так і сучасні радіотелескопи на зразок Великого масиву міліметрових антен в Атакамі[6]. На відміну від методів ядерного магнітного резонансу та спектроскопії видимого діапазону мікрохвильова спектроскопія ще не набула широкого вжитку в аналітичній хімії.
Огляд методу
Молекула в газовій фазі може вільно обертатися навколо взаємно ортогональних осей, що проходять через її центр маси. Вільне обертання неможливе у рідкій чи твердотільний фазі через міжмолекулярні сили. Обертання навколо осей пов'язане з набором квантованих рівнів енергії, значення яких залежать від моменту інерції відносно заданої осі та квантового числа. Наприклад, енергетичні рівні лінійних молекул описуються єдиним моментом інерції та єдиним квантовим числом , що визначає величину кутового моменту обертання.

Для нелінійних молекул, що є симетричними ротаторами, існують два моменти інерції, і енергія залежить від ще одного квантового числа , що визначає іншу компоненту кутового моменту відносно іншої осі[7]. Аналіз спектроскопічних даних у цьому разі допомагає визначити значення моментів інерції, а через них уточнити структуру молекули.

Для лінійної молекули аналіз спектру обертання дає значення сталої обертання[8] та момент інерції молекули, а, знаючи маси атомів, можна визначити довжину зв'язку. Для двоатомних молекул цей процес надзвичайно простий. Для лінійних молекул, що мають більше двох атомів необхідно виміряти спектри двох або більшого числа ізотопологів, таких як 16O12C32S та 16O12C34S. Це дозволяє записати систему рівнянь, з якої можна визначити довжини хімічних зв'язків[9]. Визначені довжини зв'язків будуть дещо відрізнятися від рівноважних. Така ситуація пояснюється існуванням нульових коливань, що є точкою відліку для обертальних станів, тоді як рівноважні довжини зв'язків визначаються з мінімуму потенціальної енергії. Співвідношення між сталими обертання задається формулою
- ,

де v є обертальним квантовим числом, а α is є сталою коливально-обертальної взаємодії, яку можна вирахувати, якщо значення B для двох різних обертальних станів[10].
Для інших молекул, якщо спектри можна проінтерпретувати і визначити індивідуальні переходи, можна визначити довжини зв'язків та молекулярні кути. Коли цього зробити неможливо, як для більшості асиметричних ротаторів, можна тільки підігнати спектри до трьох моментів інерції гіпотетичної молекулярної структури. Варіюючи молекулярну структуру, узгодження можна покращити. Ізотопічна заміна в цьому випадку має дуже велику цінність.
Класифікація молекулярних ротаторів
У квантовій механіці обертання молекули квантуються за значеннями кутового моменту: як енергія обертання, так і кутовий момент можуть мати тільки фіксовані значення, що відносно просто зв'язані з моментом інерції молекули. Довільна молекула має три моменти інерції , та відносно трьох взаємно ортогональних осей A, B та C, що проходять через центр маси системи. У цій статті вважається, що , тобто вісь Відповідає найменшому моментові інерції. Деякі автори визначають як вісь найвищого порядку симетрії.






Конкретні спектри енергетичних рівнів, а отже частот переходів, залежать від будови конкретної молекули. Молекули зручно розділити на чотири класи.
- Сферичні ротатори, у яких всі три моменти інерції однакові: . Серед прикладів сферичних ротаторів — тетрамери Фосфору, тетрахлорид Карбону та інші тетрагаліди, силан, гексафлорид Сульфору та інші гексагаліди. Усі ці молекули належать до кубічної груп Td або Oh.
- Лінійні молекули. В лінійних молекулах . Для більшості застосувань можна вважати . Серед прикладів молекули кисню, азоту, моноксиду Карбону, радикал гідроксилу, двоокис Карбону, ціанід Гідрогену, сульфід карбонілу, ацетилен та інші. Ці молекули належать до точкових груп C∞v або D∞h.
- Симетричні ротатори. Симетричними ротаторами є молекули з двома однаковими моментами інерції or . За означенням симетричний ротатор повинен мати вісь третього порядку. Для зручності спектроскопісти розділяють молекули на дві групи: плоскі, дископодібні, для яких та довгасті, у формі регбійного м'яча або сигари, для яких . Їхні спектри виглядають по різному, і їх легко розпізнати. Приклади включають:







- Плоскі: бензол, аміак.
- Довгасті: хлорметан, метилацетиленпропін.
- Як конкретний приклад аміак має момент інерції IC = 4.4128 × 10−47 кг м2 навколо осі третього порядку і моменти IA = IB = 2.8059 × 10−47 кг м2 навколо перпендикулярних осей. Оскільки один з моментів інерції більший, ніж два інші, молекула є плоским симетричним ротатором.[11].
- Асиметричні ротатори. У них усі три моменти інерції різні. Серед малих молекул прикладами є молекула води, діоксиду Нітрогену, у яких вісь найвище другого порядку. Більшість великих молекул теж асиметричні.
Правила відбору
Мікрохвильові та інфрачервоні спектри
Переходи між обертальними станами можна спостерігати для молекул із постійним дипольним моментом[12][13]. Як наслідок, для центрально симетричних неполярних молекул на зразок N2 або HCCH мікрохвильові спектри не спостерігаються. Тетраедральні молекули, такі як метан, що не мають дипольного моменту і мають ізотопну поляризовність, не мали б обертального спектру, якби не обертальна деформація; коли молекула обертається навколо осі третього порядку, виникає невеличкий дипольний момент, що дозволяє спостерігати слабкі лінії в мікроскопічному діапазоні[14].
Для симетричних ротаторів правила відбору дипольно дозволених обертальних переходів записується ΔK = 0, ΔJ = ±1. Оскільки вони відповідають поглинанню чи випрміненню фотона зі спіном 1, збереження кутового моменту молекули означає, що він може змінитися максимум на одиницю[15] . Більш того, квантове число K лежить у межах між +J to -J включно.
Раманівські спектри
У раманівських спектрах поглинання фотона молекулою супроводжується випромінюванням іншого. Щоб такі переходи були дозволені, поляризовність молекули повинна бути анізотропною[16]. Поляризовність є тривимірним тензором, який можна зобразити еліпсоїдом. Для сферичних ротаторів цей еліпсоїд є сферою, тож такі молекули не спостерігаються в раманівських спектрах. Інші молекули мають як стоксові так і антистоксові лінії, і приблизно однакові інтенсивності, оскільки багато обертальних рівнів термічно заселені. Правило відбору для лінійних молекул: ΔJ = 0, ±2. Причина в тому, що поляризовність двічі повертається до того ж значення за оберт[17] Значення ΔJ = 0 відповідає не молекулярному переходу, а релеївському розсіянню, коли фотон тільки змінює напрямок поширення[18].
Правила відбору для симетричних ротаторів
- ΔK = 0
- Якщо 'K = 0, то ΔJ = ±2
- Якщо K ≠ 0, то ΔJ = 0, ±1, ±2
Переходи з ΔJ = +1 R серією, тоді як переходи з ΔJ = +2 належать до S серії[18]. Оскільки в раманівських переходах беруть участь два фотони, кутовий момент молекули може змінитися на дві одиниці.
Одиниці
Одиниці обертових сталих залежать від типу вимірювань. В інфрачервоний спектроскопії зазвичай для вимірювання частоти використовують обернені сантиметри, см−1, що буквально є кількістю довжин хвилі в одному сантиметрі .З іншого боку в мікрохвильовій спектроскопії використовують гігагерци. Співвідношення між цими одиницями задається виразом


де ν — частота, λ — довжина хвилі, а c — швидкість світла.

Оскільки 1 ГГц= 109 Гц, чисельне перетворення записується
- ГГц.

Вплив коливань на обертання
Заселеність коливальних станів підкоряється розподілу Больцмана, тож стани з малою частотою коливань повинні бути помітно заселені навіть при кімнатній температурі. Оскільки для молекули, в якій збуджені коливання, момент інерції більший, обертова стала B зменшується. Як наслідок частоти обертань у кожному з коливальних станів відрізняються. Це може проявитися як сателітні лінії в обертових спектрах. Прикладом може бути ціанодіацетилен, H−C≡C−C≡C−C≡N[19].
Далі, існує фіктивна коріолісова сила між коливальними рухами ядра та обертанням (у неінерційній системі відліку). Однак, якщо коливальне квантове число не змінюється (тобто молекула має тільки одну моду коливань), вплив коливань на обертання не має великого значення, оскільки період коливань набагато менший від періоду обертання. Якщо цікавитися тільки коливальними та обертальними квантовими числами, коріолісовим зв'язком часто можна знехтувати.
Вплив обертання на коливальні спектри
Історично теорію рівнів обертання було розроблено для пояснення коливально-обертальних спектрів газів в інфрачервоному діалазоні, який використовували до того, як дослідження мікрохвильового діапазону стало практичним. У першому наближенні обертання та коливання можна вважати окремими видами руху, тож енергія обертання додається до енергії коливань. Наприклад, енергії рівнів обертання в лінійних молекулах (в наближенні жорсткого ротатора) задаються формулою

У цьому наближення, коливально-обертальні частоти переходів записуються

де та — обертальні сталі у верхньому та нижньому коливальному стані, відповідно, тоді як та є обертальними квантовими числами верхнього та нижнього рівнів. Насправді, цей вираз необхідно підправити, врахувавши ангармонійність коливань, відцентрову деформацію та коріолісові сили[20].




Для так званої R гілки спектру , тож коливання та обертання збуджуються одночасно. Для P гілки , тож квант обертальної енергії втрачається в процесі, коли збільшується квант колвальної енергії. Чисто коливальне перехід із дає Q гілку спектру. Через термічну заселеність обертальних станів P гілка дещо менш інтенсивна, ніж R гілка.



Обертальні сталі, отримані з інфрачервоних вимірювань добре узгоджуються з сталими, отриманими з мікрохвильової спектроскопії, хоча мікрохвильові вимірювання забезпечують зазвичай більшу точність.
Структура обертальних спектрів
Сферичний ротатор
Молекули, які є сферичними ротаторами, не мають дипольного моменту. Чисто обертальний спектр у них не спостерігається ні в поглилнанні ні у випромінюванні, бо фотони не мають на що діяти. Ізотропною є також поляризовність, тож чисто обертальні переходи не можуть спостерігатися і в раманівських спектрах. Однак, обертові сталі можна отримати з обертально-коливальних спетрів. Така можливість виникає, коли в збудженому стані молекула стає полярною. Наприклад, молекула метану є сферичним ротатором, але смуга, що відповідає асиметричному розтяганню C-H зв'язку, має в інфрачервоному спектрі обертальну тонку структуру. Цей спектр цікавий ще й тому, що асиметрична структура смуги свідчить про коріолісове зв'язування.
Лінійні молекули
Жорсткий ротатор є добрим віправним пунктом для конструювання моделі молекули, що обертається. Припускається, що атоми є точковими масами, сполученими стрижнями незмінної довжини. Лінійна молекула лежить на єдиній осі, і кожен атом рухається на поверхні сфери навколо центру інерції. Два ступені обертальної вільності відповідають сферичним координатам θ та φ, що задають положення осі молекули, а квантові стани індексуються двома квантовими числами J та M. J визначає величину обертального кутового моменту, а M — його складові відносно фіксованої осі, наприклад, заданої магнітним полем. Без зовнішніх полів енергія залежить лише від J. У моделі жорсткого ротатора енергія рівнів обертання молекули F(J) задається як

де — обертальна стала молекули, що пов'язана з її моментом інерції. Момент інерції лінійної молекули перпендикулярний до осі обератння і є єдиним: , тож



Для двоатомної молекули

де m1 та m2 — маси атомів, а d — відстань між ними.
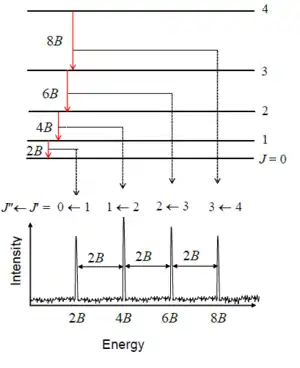
Правила відбору вимагають, щоб при випромінюванні або поглинанні обертове квантове число змінювалося на одиницю; тобто . Тому положення ліній в обертальному спектрі задаються формулою


де позначає нижній рівень, а позначає верхній рівень із тих, що беруть участь у переході.


Діаграма ілюструє обертальні переходи, підпорядковані правилу відбору =1. Штриховими лініями показано як ці переходи вписуються в експериментальну структуру спектру. Суміжні переходи в спектрі розділені величиною 2B.


Інтенсивності обертальних ліній
Імовірність переходу є найважливішим фактором, що впливає на інтенсивність експериментальних обертальних ліній. Ця ймовірність пропорційна заселеності початкового стану, з якого відбувається перехід. Заселеність обертального стану залежить від двох факторів. Один із них — кількість молекул в збудженому стані з квантовим числом J, відносно до кількості молекул в основному стані, NJ/N0 задається розподілом Больцмана як
- ,

де k — стала Больцмана, а T — абсолютна температура. Цей фактор зменшується зі зростанням J. Другий фактор — виродженість обертального стану, що дорівнює 2J+1. Цей фактор зростає зі зростанням J. Враховуючи обидва[21]
- заселеність

Максимум відносної інтенсивності припадає на[22][23]

Відцентрове спотворення
Коли молекула обертається, відцентрові сили намагаються розтягнути атоми в різні сторони. Як наслідок, момент інерції молекули збільшується, а отже значення зменшується в порівнянні зі значенням, розрахованим для жорсткого ротатора. Щоб врахувати це, до енергії двоатомної молекули дописують додатковий корекційний член[24]

де — певна стала.

Тому положення лінії обертової моди змінюється до

Як наслідок, відстань між лініями не стала, як у наближенні жорсткого ротатора, а зменшується зі збільшенням обертального квантового числа.
Наведений визраз витікає з припущення, що коливання в молекулі підкоряються гармонічному законові руху. В гармонічному наближенні відцентрова стала виводиться як

де k — силова стала коливального руху. Звідси слідує співвідношення між та

де — частота коливань.

Якщо враховувати ангармонічність, у виразі для енергії рівнів та для положення ліній з'являються члени вищого степеня J[24]. Яскравим прикладом є флуорид гідрогену, спектр якого було підігнано до членів із [J(J+1)]5'[25].
Молекула кисню
Електричний дипольний момент молекули кисню дорівнює нулю, але вона є парамагнітною з двома неспареними електронами, тож існують дозволені магнітні переходи, які можна спостерігати в мікрохвильовому діапазоні. Одиничний спін молекули має три просторові орієнтації щодо кутового моменту обертання K, тож кожен обертальний рівень розщеплюється на три стани з J = K + 1, K та K - 1. Виникає триплет, який називають триплетом p-типу. Різниця енергій між станами з сусідніми J в кожному з цих триплетів дорівнює приблизно 2 см−1 (60 ГГц). Винятком є різниця J = 1←0, що становить приблизно 4 см−1. Правила відбору для магнітних дипольних переходів дозволяють переходи між послідовними членами триплетів (ΔJ = ±1), так що для кожного значення обертального кутового моменту K існує два дозволені переходи. Ядро 16O має нульовий ядерний спін, тож міркування симетрії вимагають, щоб K пробігало тільки непарні значення[26][27].
Симетричний ротатор
Для симетричного ротатора квантове число J асоційоване з повним кутовим моментом молекули. Для заданого J існує 2J+1-кратна виродженість за квантовим числом M, що пробігає значення +J ...0 ... -J. Третє квантове число K пов'язане з обертанням навколо головної осі молекули. За відсутності електричного поля енергія обертання залежить тільки від J та K, і в наближенні жорсткого ротатора енергія кожного обертового стану задається формулою

де , для видовженої молекули і для сплюснутої молекули.



Це визначає частоти переходів як

як і для лінійних молекул[28].
З поправкою першого порядку на відцентрове спотворення частоти переходів стають

Член DJK знімає виродженість щодо різних значень K, притаманну наближенню жорсткого ротатора[29].
Асиметричний ротатор
Як і раніше повний кутовий момент позначається J. Оскільки незалежних моментів інерції три, треба враховувати два інші квантові числа, але прості вирази для спектру асиметричного ротатора вивести не можна. Спектр отримують окремо для кожного J діагоналізацією матриці. Існують формули для тих молекул, чия форма близька до форми симетричногго ротатора[30].
Важливим прикладом несиметричного ротатора є молекула води. Вона має інтенсивний чисто ротаційний спектр в далекому інфрачервоному діапазоні, нижче від 200 см−1. Саме через це інфрачервоні спектроскопи потрібно очистити від атмосферної водяної пари або за допомогою сухого газу або викачуванням. Спектр води детально проаналізовано[31].
Квадрупольне розщеплення
Коли спін ядра I більший ніж 1/2, воно має квадрупольний момент. У цьому разі взаємодія ядерного спіну з кутовим моментом обертання призводить до розщеплення рівнів обертової енергії. Коли J обертового стану більше, ніж I виникає 2I+1 рівнів; але коли J менше, ніж I виникає 2J+1 рівнів. Це явище є прикладом надтонкого розщеплення. Наприклад, для 14N (I = 1) в HCN усі рівні з J > 0 розщепляються на 3. Енергії підрівнів пропорційні квадрупольному моменту та функції від F і J. де F = J+I, J+I-1, ..., 0, ... |J-I|.
Отже, спостереження ядерного квадрупольного розщеплення дозволяє визначити величину ядерного квадрупольного моменту[32]. Цей метод є альтернативою спектроскопії ядерного квадрупольного резонансу. Правила відбору для обертальних переходів стають[33]

Ефекти Штарка та Зеємана
У статичному зовнішньому електричному полі 2J+1 разове виродження кожного з обертових станів частково знімається, що є прикладом ефекту Штарка. Зокрема, кожен із енергетичних рівнів лінійної молекули розщеплюється на J+1. Величина розщеплення залежить від квадрату напруженості електричного поля та квадрату дипольного моменту молекули[34]. В принципі це дозволяє з великою точністю визначити значення дипольних моментів молекул. Прикладом може бути сульфід карбонілу, OCS, з μ = 0,71521 ± 0,00020 Д. Однак, з огляду на те, що розщеплення залежить від μ2, орієнтацію диполя потрібно знаходити з квантовомеханічних міркувань[35].
Аналогічне зняття виродження відбувається з парамагнітною молекулою в магнітному полі завдяки ефекту Зеємана. Більшість сполук, що досліджуються в газовому стані діамагнітні. Винятком є молекули з неспареним електроном, такі як оксид нітрогену NO, діоксид нітрогену, NO2, деякі оксиди хлору та радикал гідроксилу. Ефект Зеємана спостерігався на молекулі кисню O2[36].
Ротаційна Раман-спектроскопія
Обертальні переходи молекули можна також спостерігати методом Раман-спектроскопії. Обертальні переходи дозволені в раманівських спектрах для будь-якої молекули, що має анізотропічну поляризовність, а це означає будь-які молекули крім симетричних ротаторів. Звідси слідує, що в розсіянні можна спостерігати обертальні переходи молекул, які не мають постійного дипольного моменту, тоді як у поглинанні чи випромінюванні їх бачити не можна. Раман-спектри з високим розділенням можна отримати, застосовуючи інфрачервоні Фур'є-спектрометри. Прикладом може бути спектр 15N2. У ньому видно вплив ядерного спіну, що проявляється в зміні інтерсивностей сусідніх ліній 3:1. З даних було зроблено висновок, що довжина зв'язку дорівнює 109,9985 ± 0,0010 пм[37]
Інструменти та методи
Переважна більшість сучасних спектрометрів є комбінацією комерційних та саморобних складових, що використовуються для конкретних потреб. Інструменти можна категоризувати відповідно до загальних принципів дії. Хоча обертальні переходи можна бачити в дуже широкому спектральному діапазоні, існують фундаментальні фізичні обмеження на діапазони окремих засобів. Часто непрактично й дорого переходити до зовсім іншого діапазону частот. Інструменти та засади їхньої дії, описані нижче, здебільшого відповідають мікрохвильовій спектроскопії на частотах між 6 та 24 ГГц.
Комірка поглинаня та модуляція Штарка
Найлегше мікрохвильовий спетрометр сконструювати, використовуючи джерело мікрохвильового випромінювання, комірку поглинання, в яку вводять призначений для вимірювання газ, та детектор, такий як супергетеродинний приймач. спектр можна отримати, змінюючи частоту джерела й вимірюючи інтенсивність сигналу, що пройшов через комірку. Коміркою може бути просто секція хвилеводу. Важливим різновидом методу є застосування змінного струму на електродах в комірці поглинання, що призоводить до модуляції частоти обертальних переходів. Це називають штарковою модуляцією, яка дозволяє використання чутливого до фази детектування й покращує чутливість. Спектоскопія поглинання дозволяє досліджувати термодинамічно стабільні зразки при кімнатній температурі.
Перше дослідження мікрохвильового спектру молекули NH3 здійснили Клітон та Вільямс у 1934[38]. Пізніші експерименти використовували потужні джерела мікрохвильового випромінювання на зразок клістрона, багато з яких були розроблені для радарів під час Другої Світової війни. Число експериментів з мікрохвильвої спектроскопії збільшилося одразу ж після війни. До 1948 року Волтер Горді зміг написати огляд результатів понад 100 наукових робіт[39]. Комерційний[40] спектрометр поглинання, який розробила компанія Hewlett Packard в 1970-х, свого часу широко використовувався в фундаментальних дослідженнях. Наразі більшість лабораторій світу використовують або спектрометр Болла-Флайгара або чирпований (зі змінною миттєвою частотою) Фур'є-спектрометр (FTMW).
Мікрохвильовий Фур'є-спектрометр (FTMW)
Теоретичні засади[41] Фур'є-спектроскопії в мікрохвильовому діапазоні аналогічні теорії Фур'є-спектроскопії ядерного магнітного резонансу (FT-NMR). Еволюція системи описується оптичними рівняннями Блоха. Спочатку запускають короткий (зазвичай тривалістю 0-3 мікросекунди) імпульс в резонансі з вхідною хвилею випромінювання. Ті молекули, що поглинають енергію імпульсу починають обертатися когерентно в фазі з вхідним сигналом. Деактивація поляризаційного імпульсу супроводжується мікрохвильовим випромінюванням, що відповідає декогеренції молекулярного ансамблю. Цей вільний індукційний розпад відбувається за проміжок часу 1-100 мікросекунд, залежно від налаштувань інструменту. Слідуючи піонерським роботам Дікке 1950-х[42], перший FTMW-спектрометр сконструювали Еккерз та Флайгар у 1975[43].
Фур'є-спектрометр Болла-Флайгара
Болл, Кемпбеллл, Кінан та Флайгар показали, що метод мікрохвильової Фур'є-спектроскопії можна використовувати у комірці вільного простору, тобто у вакуумній камері з резонатором Фабрі-Перо[44]. Ця методика дозволила вивчати зразок уже через мілісекунди після швидкого охолодження до темпетатури кількох кельвінів у соплі газового струмення, що розширюється. Це стало революційною розробкою, бо охолодження молекул до низьких температур дозволяє зібрати молекули на найнижчих обертових рівнях енергії. Разом із використанням резонатора Фабрі-Перо це принесло значне збільшення чутливості та роздільної здатності спектрометрів разом із зменшенням складності досліджуваних спектрів. Стало можливим виокремлювати й вивчати дуже слабко зв'язані молекули, бо за таких низьких температур їм нізвідки взяти енергію, щоб розлетітися на фрагменти чи вступити в хімічні реакції. Піонером в дослідженні слабко зв'язаних сполук став Вільям Клемперер. Тоді як резонатор Фабрі-Перо у спектрометрі Балла-Флайгара можна налаштувати в резонанс із будь-якою частотою від 6 до 18 ГГц, ширина діапазону окремих вимірювань обмежена приблизно 1 МГц. У зовнішньому посиланні[45] є анімація, що демонструє принцип дії цього популярного в мікрохвильовій спектроскопії інструмента.
Чирпований Фур'є-спектрометр
Звернувши увагу на той факт, що техніка оцифрування та відповідна електроніка сильно розвинулись з часів винаходу метода Фур'є-спектроскопії, Брукс Пейт із університету Вірджинії[46] розробив спектрометр[47], що зберігає багато цінних рис спектрометра Болла-Флайгара, водночас внісши суттєві інновації:
- використання швидкісного (>4 GS/с) генератора хвильового імпульсу, що створює чирпований поляризаційний сигнал, який пробігає частотний діапазон до 12 ГГц за мікросекунду й менше;
- використання швидкісного осцилографа (понад 40 GS/с), що оцифровує сигнал декогеренції молекул та записує його Фур'є-спектр. Виник інструмент, що не тільки дозволяє вивчати молекули зі слабким зв'язком, але й значно розширює частотний діапазон (до 12 ГГц) у порівнянні зі спектрометром Болла-Флайгара.
Модифіковані варіанти першого чирпованого Фур'є-спектрометра сконструювали численні групи в Сполучених Штатах, Канаді й Європі[48][49]. Інструмент забезпечує широкий частотний діапазон, що доповнює чутливість та роздільну здатність розробки Болла-Флайгара.
Виноски
- Спектр було виміряно упродовж пари годин за допомогою пульсуваного мікрохвильового спектрометра з Фур'є перетворювачем у Бристольському університеті
- Gordy, W. (1970). У A. Weissberger. Microwave Molecular Spectra in Technique of Organic Chemistry IX. New York: Interscience.
- Nair, K.P.R.; Demaison, J.; Wlodarczak, G.; Merke, I. (236). Millimeterwave rotational spectrum and internal rotation in o-chlorotoluene. Journal of Molecular Spectroscopy 237 (2): 137–142. Bibcode:2006JMoSp.237..137N. doi:10.1016/j.jms.2006.03.011.
- Cheung, A.C.; Rank, D.M.; Townes, C.H.; Thornton, D.D.; Welch, W.J. (1968). Detection of NH3 molecules in the interstellar medium by their microwave emission spectra. Physical Review Letters 21 (25): 1701–5. Bibcode:1968PhRvL..21.1701C. doi:10.1103/PhysRevLett.21.1701.
- Ricaud, P.; Baron, P; de La Noë, J. (2004). Quality assessment of ground-based microwave measurements of chlorine monoxide, ozone, and nitrogen dioxide from the NDSC radiometer at the Plateau de Bure. Ann. Geophys 22: 1903–15. Bibcode:2004AnGeo..22.1903R. doi:10.5194/angeo-22-1903-2004.
- Astrochemistry in Virginia. Процитовано 2 грудня 2012.
- Atkins & de Paula, 2006, p. 444
- Ця стаття використовує звичну серед спектроскопістів конвенцію виражати сталу обертання в см−1.
- Для симетричного ротатора значення двох моментів інерції дозволяють визначити два параметри. Кожен додатковий ізотополог дає інформацію про ще один параметр молекули. Для несиметричних ротаторів кожен ізотополог дає інформацію про щонайбільше три молекулярні параметри.
- Banwell & McCash, 1994, p. 99
- Moment of inertia values from Atkins & de Paula, 2006, p. 445
- Hollas, 1996, p. 95
- Такі переходи називають дипольний дозволеними. Дозволеними можуть бути інші переходи з участю квадрупопів, октаполів гексадекаполів тощо, але вони мають дуже малу інтенсивність, тож спостерігати їх дуже важко. Магнітні дипольно дозволені переходи можуть відбуватися в парамагнітних молекулах, таких як молекула кисню та оксид Нітрогену NO.
- Hollas, 1996, p. 104 наводить частину ротаційного спектру силану
- Atkins & de Paula, 2006, p. 447
- Hollas, 1996, p. 111
- Atkins & de Paula, 2006, pp. 474–5
- Banwell & McCash, 1994, Section 4.2, p. 105, Pure Rotational Raman Spectra
- Alexander, A. J.; Kroto, H. W.; Walton, D. R. M. (1967). The microwave spectrum, substitution structure and dipole moment of cyanobutadiyne. J. Mol. Spectrosc 62: 175–180. Bibcode:1976JMoSp..62..175A. doi:10.1016/0022-2852(76)90347-7. Illustrated in Hollas, 1996, p. 97
- Banwell & McCash, 1994, p. 63.
- Banwell & McCash, 1994, p. 40
- Atkins & de Paula, 2006, p. 449
- Це значення J відповідає максимуму заселеності, якщо її розглядати як функцію неперервного J. Однак, оскільки дозволені тільки цілі значення, максимальна лінія має найближче значенння цілого J.
- Banwell & McCash, 1994, p. 45
- Jennings, D.A.; Evenson, K.M; Zink, L.R.; Demuynck, C.; Destombes, J.L.; Lemoine, B; Johns,J.W.C. (April 1987). High-resolution spectroscopy of HF from 40 to 1100 cm−1: Highly accurate rotational constants. Journal of Molecular Spectroscopy 122 (2): 477–480. Bibcode:1987JMoSp.122..477J. doi:10.1016/0022-2852(87)90021-X.pdf
- Strandberg, M. W. P.; Meng, C. Y.; Ingersoll, J. G. (1949). The Microwave Absorption Spectrum of Oxygen. Phys.Rev. 75 (10): 1524–8. Bibcode:1949PhRv...75.1524S. doi:10.1103/PhysRev.75.1524.pdf
- Krupenie, Paul H. (1972). The Spectrum of Molecular Oxygen. J. Phys. Chem. Ref. Data 1, 423 (1972) 1 (2): 423–534. Bibcode:1972JPCRD...1..423K. doi:10.1063/1.3253101.
- Hollas, 1996, p. 101
- Hollas, 1996, p. 102 shows the effect on the microwave spectrum of H3SiNCS.
- Hollas, 1996, p. 103
- Hall, Richard T.; Dowling, Jerome M. (1967). Pure Rotational Spectrum of Water Vapor. J. Chem. Phys. 47 (7): 2454–61. Bibcode:1967JChPh..47.2454H. doi:10.1063/1.1703330. Hall, Richard T.; Dowling, Jerome M. (1971). Erratum: Pure Rotational Spectrum of Water Vapor. J. Chem. Phys. 54 (11): 4968. Bibcode:1971JChPh..54.4968H. doi:10.1063/1.1674785.
- Simmons, James W.; Anderson, Wallace E.; Gordy,Walter (1950). Microwave Spectrum and Molecular Constants of Hydrogen Cyanide. Phys. Rev. 77: 77–79. Bibcode:1950PhRv...77...77S. doi:10.1103/PhysRev.77.77.
- Chang, Raymond (1971). Basic Principles of Spectroscopy. McGraw-Hill. p139
- Hollas, 1996, p. 102 наводить рівняння для двоатомної молекули та симетричного ротатора
- Hollas, 1996, p. 102
- Burkhalter, James H.; Roy S. Anderson; William V. Smith; Walter Gordy (1950). The Fine Structure of the Microwave Absorption Spectrum of Oxygen. Phys. Rev. 79 (4): 651–5. Bibcode:1950PhRv...79..651B. doi:10.1103/PhysRev.79.651.
- Hollas, 1996, p. 113 ілюструє спектр 15N2, отриманий розсіянням випромінювання аргонового йонного лазера з частотою 476,5 нм.
- Cleeton, C.E.; Williams, N.H. (1934). Electromagnetic waves of 1.1 cm wave-length and the absorption spectrum of ammonia. Physical Review 45 (4): 234–7. Bibcode:1934PhRv...45..234C. doi:10.1103/PhysRev.45.234.
- Gordy, W. (1948). Microwave spectroscopy. Reviews of Modern Physics 20 (4): 668–717. Bibcode:1948RvMP...20..668G. doi:10.1103/RevModPhys.20.668.
- June 1971, Hewlett Packard Journal. Процитовано November 2012.
- Schwendemann, R.H. (1978). Transient Effects in Microwave Spectroscopy. Annu. Rev. Phys. Chem. 29: 537–558. Bibcode:1978ARPC...29..537S. doi:10.1146/annurev.pc.29.100178.002541.
- Dicke, R.H.; Romer, R.H. (1955). Pulse Techniques in Microwave Spectroscopy. Rev. Sci. Inst. 26 (10): 915–928. Bibcode:1955RScI...26..915D. doi:10.1063/1.1715156.
- Ekkers, J.; Flygare, W.H. (1976). Pulsed microwave Fourier transform spectrometer. Rev. Sci. Inst. 47 (4): 448–454. Bibcode:1976RScI...47..448E. doi:10.1063/1.1134647.
- Balle, T.J.; Campbell, E.J.; Keenan, M.R.; Flygare, W.H. (1980). A new method for observing the rotational spectra of weak molecular complexes: KrHCl. J. Chem. Phys. 72 (2): 922–932. Bibcode:1980JChPh..72..922B. doi:10.1063/1.439210.
- Jager, W. Balle-Flygare FTMW spectrometer animation.
- Web page of B.H. Pate Research Group, Department of Chemistry, University of Virginia. Процитовано November 2012.
- Brown, G.G.; Dian, B.C.; Douglass, K.O.; Geyer, S.M.; Pate, B.H. (2006). The rotational spectrum of epifluorohydrin measured by chirped-pulse Fourier transform microwave spectroscopy. J. Mol. Spectrosc. 238: 200–212. Bibcode:2006JMoSp.238..200B. doi:10.1016/j.jms.2006.05.003.
- Grubbs, G.S.; Dewberry, C.T.; Etchison, K.C.; Kerr, K.E.; Cooke, S.A. (2007). A search accelerated correct intensity Fourier transform microwave spectrometer with pulsed laser ablation source. Rev. Sci. Inst. 78 (9): 096106. Bibcode:2007RScI...78i6106G. doi:10.1063/1.2786022.
- Wilcox, D.S.; Hotopp, K.M.; Dian, B.C. (2011). Two-Dimensional Chirped-Pulse Fourier Transform Microwave Spectroscopy. J. Phys. Chem. A 115 (32): 8895–8905. doi:10.1021/jp2043202.