Нейрофіброматоз І типу
Нейрофіброматоз І типу (англ. Neurofibromatosis I type, NF1, нейрофіброматоз 1-й тип; застаріле — хвороба Реклінггаузена, англ. Recklinghausen disease; периферичний нейрофіброматоз) — один з типів нейрофіброматозу. Є найпоширенішою моногенною спадковою хворобою, яка призводить до виникнення пухлин у людини. NF1 є офіційним міжнародним генетичним символом нейрофіброматозу цього типу.
| Нейрофіброматоз І типу / Хвороба Реклінгаузена | |
|---|---|
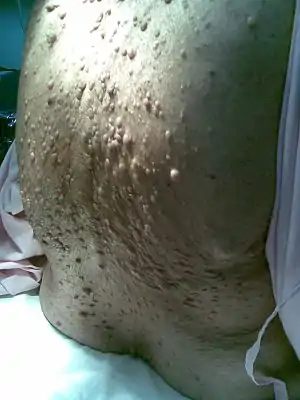 Типові прояви нейрофіброматозу І типу на спині та сідницях хворої — підшкірні вузли, які являють собою пухлини Типові прояви нейрофіброматозу І типу на спині та сідницях хворої — підшкірні вузли, які являють собою пухлини | |
| Спеціальність | медична генетика і неврологія[1] |
| Частота | 0.026998%, 0.0128%, 0.0217%, 0.0241%, 0.0457%, 0.0149%, 0.02691% |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | LD2D.10 |
| МКХ-10 | Q85.0 |
| OMIM | 162200 |
| DiseasesDB | 8937 |
| MedlinePlus | 000847 |
| eMedicine | radio/474 |
| MeSH | D009456 |
| | |
Історичні відомості
Хоча перші описи відомі ще з I століття, однак медичний опис хвороби було зроблено у другій половині XIX століття низкою дослідників, але найбільше відомий той опис хвороби, що зробив у 1882 році учень Рудольфа Вірхофа Фрідріх фон Реклінггаузен, на честь якого хвороба отримала свою першу епонімічну назву[2].
Актуальність
Трапляється з однаковою частотою як в осіб чоловічої статі, так і жіночої, у 1 з 3500 новонароджених. Станом на 2015 рік щонайменше 100 тисяч людей в США і 15 тисяч у Великій Британії мали цю хворобу. Ризик народження дитини з нейрофіброматозом від хворого складає від 50 % (у випадку гетерозиготи) до 100 % (в разі гомозиготи). Темп мутацій гена NF1 є одним з найвищих серед усіх відомих спадкових захворювань людини — сягає 6,5 х 10−5 гамет на покоління, чи близько 1 на 10 000 гамет.
Етіологія
Достеменно невідома.
Патогенез
Локус генів, поломка яких призводить до розвитку нейрофіброматозу цього типу, розташований на довгому плечі 17 хромосоми (17q11.2). Він складається з 400 тисяч нуклеотидних пар, є дуже протяжним і складно організованим. У ньому міститься інформація, відповідальна за синтез одного зі складових мієліну — глікопротеїну, нейрофіброміну та інших білків. При цьому можливі у цьому локусі різні типи мутацій і поломок — транслокації, делеції, інверсії і точкові мутації. Характер мутацій вельми специфічний: більше 80 % з них ведуть до синтезу нефункціонального «усіченого» білка або до повної відсутності транскрипту (нонсенс-мутації, мутації в сайтах сплайсингу, делеції та інсерції із зсувом рамки, великі делеції, що охоплюють увесь ген або його значну частину).
Нейрофібромін є цитоплазматичним білком, що бере участь в інактивації білків-промоторів — білка RAS і його аналогів, забезпечуючи таким чином динамічний контроль росту клітин. Ген NF1 є одним з головних генів-супресорів пухлинного росту для приблизно половини тканин організму, в першу чергу, нейроектодермального походження, проліферацію яких визначає система білків RAS. Нейрофібромін також надає вплив на вміст у клітині аденозинмонофосфату (АМФ). АМФ в свою чергу опосередковано гальмує процеси клітинного поділу.
При пошкодженні гена NF1 в одній з хромосом 17 пари половина синтезованого нейрофіброміну стає дефектною, і відзначається зміщення рівноваги росту клітин в бік проліферації. Той неушкоджений алельний ген NF1, що знаходиться в парній хромосомі, забезпечує синтез нормального нейрофіброміну. Виразність клінічних проявів нейрофіброматозу визначається станом загального протипухлинного імунітету і може варіювати в широких межах. Виникають як доброякісні новоутворення, так і в разі втрати через мутацію алельного нормального гена НФ1 відбувається стрімкий і бурхливий неконтрольований ріст клітин, розвивається злоякісна пухлина — найчастіше нейрофібросаркома або нейробластома. Імовірність їх виникнення сягає 15 %. Імовірність розвитку асоційованої з цим типом нейрофіброматозу злоякісної пухлини перевищує таку в популяції в сотні разів, зокрема, стосовно мієлолейкоза в 200—500 разів.
Клінічні ознаки
Хвороба проявляється множинними нейрофібромами по ходу периферичних нервів, які являють собою болючі округлі вузлики у товщі шкіри, що варіюють за своїми розмірами (від просяного зерна до 7 см в діаметрі) та локалізацією, не зміщується повздовж, а тільки в поперечному напрямку разом з нервовим стовбуром. Найчастіше перші видимі нейрофіброми з'являються у передпубертатний або пубертатний період. До 30-річного віку відзначається неухильне повільне збільшення нейрофібром, особливо помітне під час статевого дозрівання пацієнта, а також під час вагітності у жінок. Після чого зростання нейрофібром певною мірою стабілізується.
У деяких випадках нейрофіброматозні зміни можуть носити досить обмежений сегментарний характер (зокрема, медіастинальні нейрофіброми спільно з пухлинами у відповідному шкірному сегменті), але набагато частіше вони є генералізованими — пухлини виникають одночасно на тулубі, шиї, голові, кінцівках.
Нерідко поява нейрофібром супроводжується слоновістю уражених ділянок тіла і внутрішніх органів. Плексиформні нейрофіброми частіше поодинокі, але при цьому можуть досягати великих розмірів. Вони представлені розростанням тканин нерва з нормальних навколишніх тканин. Хоча частота виникнення плексиформних нейрофібром сягає лише 5 %, але вони доставляють великі проблеми при появі. Найчастіше вони починають розвиватися ще до народження дитини і стають симптомними до дворічного віку.
До пухлиноподібних розростань на шкірі відносять папіломи. Вони зустрічаються набагато рідше, ніж пухлини в підшкірній клітковині. Сегментарний нейрофіброматоз пов'язують з соматичними мутаціями на певній стадії ембріогенезу, що призводять до залучення обмеженого клону клітин і соматичному мозаїцизму.
Типові для цього нейрофіброматозу плоскі пігментні плями «кави з молоком» і «веснянкуватих грон» на шкірі, які зазвичай з'являються до дворічного віку, а також вузликів Ліша — пігментних плям на райдужці ока, що виявляють при офтальмологічному огляді за допомогою щілинної лампи. Виявлення вузликів Ліша підвищується з віком пацієнта: у віці до 4 років їх виявляють у 22 % випадків, тоді як у старших 20 років — до 95 %. Як правило, плями «кава з молоком» і вузлики Ліша не несуть безпосередньої небезпеки для здоров'я людини, але зливні пігментні плями на шкірі можуть доставляти незручність як косметичний дефект. Іноді пігментні шкірні плями є єдиним клінічним проявом, так як невеликі нейрофіброми не завжди вдається знайти, особливо у дітей. Пігментні плями дуже різноманітні за кількістю, розмірами, локалізації, забарвленням. У третини хворих відбувається характерний розвиток пігментних плям в пахвових та пахових ділянках, так звані знаки Кроу.
Незважаючи на периферичний характер цього типу нейрофіброматозу, у частини хворих може спостерігатися залучення центральної і периферичної нервової системи з формуванням пухлин іншої гістологічного генезу — астроцитом і гліом зорових шляхів, епендими, менінгіом, шванном, спінальних нейрофібром. Оптична гліома — доброякісна пухлина зорового нерва, вона рідко зустрічається у дітей молодшого віку, частіше дебютує в десятирічному віці поступовим зниженням зору. Також можуть виявлятися пухлини іншої локалізації, включаючи пухлину наднирників феохромоцитому. Мутація гена NF1 може бути причиною мієлодиспластичного синдрому та рідкісного виду лейкозу — ювенільного мієломоноцитарного лейкозу (англ. Juvenile myelomonocytic leukemia — JMML), що є патогномонічною ознакою, якщо відбувається у дітей молодше двох років.
Для цього типу нейрофіброматозу характерні також:
- у 50 % випадків когнітивні порушення різного ступеня (від легких до виражених), частіше в поєднанні з не сильним або помірним зниженням IQ, утрудненням в освоєнні письма, читання, математики;
- ендокринні розлади — порушення росту і статевого дозрівання;
- зміни скелета — сколіоз, деформація грудної клітки, спондилолістез, відсутність заростання дужок хребців, краніовертебральні аномалії, асиметрія черепа, псевдоартроз;
- епілептичні напади;
- часті переломи кісток кінцівок, які важко піддаються лікуванню (довго зростаються) і вимагають втручання фахівця (ортопеда).
У дітей при цьому може бути знижений м'язовий тонус, порушена координація рухів. У них великий розмір черепа (окружність голови) — понад 4 стандартних відхилень, ніж зазвичай в цьому віці. Вони можуть бути нижчими на зріст, ніж очікується, в порівнянні з іншими членами сім'ї. Такі діти можуть бути малоініціативними і менш емоційними в порівнянні зі здоровими однолітками. Близько 12 % пацієнтів, хворих на цей тип нейрфіброматозу, мають фенотип, характерний для синдрому Нунан (англ. Noonan): гипертелоризм, антимонголоїдний розмір очей, низько посаджені вуха, шийний птерігум, стеноз легеневої артерії.
Діагностика
Клінічними критеріями є наявність:
- шести чи більше світло-коричневих пігментних плям на шкірі («café au lait spots»);
- принаймні двох нейрофібром;
- щонайменше двох нодулярних (вузлових) утворень на райдужці очей (вузлики Ліша);
- аномалії хребта — сколіозу.
Генетична діагностика
Використовують як пряму ДНК-діагностику, так і непряму. Даний метод може бути доповнений цілим рядом інших традиційних технологій мутаційного скринінгу (зокрема, гетеродуплексний аналіз, градієнтний денатураційний гель-електрофорез, блот-гібридизація, пряме секвенування окремих екзонів гена тощо). Непряма ДНК-діагностика значно простіше і економніше в порівнянні з прямою, тому вона не втратила свого значення і сьогодні. Тісне зчеплення використаних маркерів з геном NF1 дозволяє проводити непряму ДНК-діагностику цього типу нейрофіброматозу з точністю 98 %.
Детальніші відомості з цієї теми ви можете знайти в статті Нейрофіброматоз.
Лікування
На даний час не розроблено ефективної терапії цього типу нейрофіброматозу. При зниженні здатності до навчання і когнітивних порушеннях рекомендується навчання дітей і підлітків або вдома, або в спецшколах і проведення постійної соціальної реабілітації пацієнтів. Пухлини є причиною больового синдрому і зниження функцій. При вираженому больовому синдромі призначають нестероїдні протизапальні препарати, неопіодні та опіоїдні анальгетики, трициклічні антидепресанти, габапентин. Ортопедичні операції можливі за наявності кісткових деформацій, сколіозу. Хірургічні операції також проводять при наявності болючих нейрофібром, ліпом або папілом великих розмірів, а також при розташуванні пухлин в областях постійної травматизації або пухлин, які спричинюють косметичний дефект. Променева терапія і хіміотерапія проводиться у випадках малігнізації пухлин.
Примітки
- Bednařík J., Ambler Z., Růžička E. Klinická neurologie: část speciální — ISBN 978-80-7387-389-9
- Whonamedit?- A dictionary of medical eponyms. Friedrich Daniel von Recklinghausen (англ.)
Джерела
- Неврологія: національний підручник / I.А. Григорова, Л.I. Соколова, Р. Д. Герасимчук та ін.; за ред. I.А. Григорової, Л.I. Соколової. — К.: ВСВ «Медицина», 2015. — 640 с. + 32 с. кольор. вкл.
- Evans, Rosalie E. Ferner, Susan M. Huson, D. Gareth R. (2011). Neurofibromatoses in clinical practice. London: Springer. p. 1. ISBN 978-0-85729-628-3 (англ.)
- Boyd, Kevin P.; Korf, Bruce R.; Theos, Amy (July 2009). «Neurofibromatosis type 1». Journal of the American Academy of Dermatology. 61 (1): 1–14. (англ.)
- Neurofibromatosis type 1. Genetics Home Reference. 2015-10-05. (англ.)
- Choices, NHS. «Neurofibromatosis type 1 — Causes — NHS Choices». (англ.)
- «Neurofibromatosis 1: Current Issues in Diagnosis, Therapy, and Patient Management», by David Viskochil MD PhD, Mountain States Genetic Foundation, Denver 2010 (англ.)
- «Current Therapies for Neurofibromatosis Type 1», by Laura Klesse MD PhD, Mountain States Genetic Foundation, Denver 2010 (англ.)
- Abernathy CR, Rasmussen SA, Stalker HJ, Zori R, Driscoll DJ, Williams CA, Kousseff BG, Wallace MR. NF1 mutation analysis using a combined heteroduplex/SSCP approach. // Hum Mutat.. — 1997. — Т. 9. — С. 548-54. (англ.)
- Kluwe L., Tatgiba M., Fünsterer C., Mautner V.-F. NF1 mutations and clinical spectrum in patients with spinal neurofibromas // J Med Genet. — 2003. — Т. 40. — С. 368—371. (англ.)
- Шнайдер Н. А. Нейрофиброматоз 1-го типа: этиопатогенез, клиника, диагностика, прогноз // Международный неврологический журнал. — 2007. — № 5 (15). (англ.)