Синдром де Тоні — Дебре — Фанконі
Синдро́м де Тоні — Дебре́ — Фанко́ні[1] (первинний ізольований синдром Фанконі, глюкозо-фосфат-аміновий діабет) — вроджене захворювання, успадковується за аутосомно-рецесивним типом[2]. Відбувається комплекс біохімічних і клінічних проявів ураження проксимальних ниркових канальців з порушенням канальцевої реабсорбції фосфату, глюкози, амінокислот і бікарбонату[3]. Одне з рахітоподібних захворювань.
| Синдром де Тоні — Дебре — Фанконі | |
|---|---|
| Спеціальність | нефрологія і ендокринологія |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | GB90.42 |
| МКХ-10 | E72.0 |
| OMIM | 613388, 615605 і 134600 |
| DiseasesDB | 11687 |
| eMedicine | ped/756 |
| MeSH | D005198 |
| SNOMED CT | 40488004 |
Історичні відомості
Першим цей синдром описав 1933 року італійський педіатр Д. де Тоні, виділивши гіпофосфатемію при цьому.[4]. 1934 року французький педіатр Р. Дебре описав у дитини з карликовістю і рахітом глюкозурію і альбумінурію[5] Цю тубулопатію зрештою ідентифікував швейцарський педіатр Г. Фанконі у 1936 році серед раніше описаних іншими дослідниками окремих частин захворювання.[6] Тому на сьогодні вважають означати епонімічну приналежність цього синдрому саме в такому порядку «де Тоні — Дебре — Фанконі».[1]
Етіологія
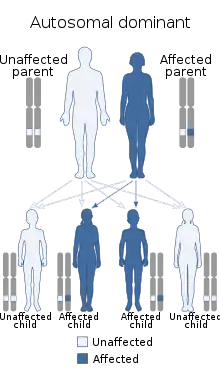
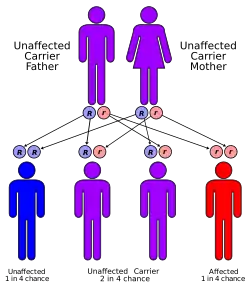
Найчастіше синдром є компонентом інших спадкових хвороб: цистиноз, тирозинемія типу I, галактоземія, хвороба Вільсона, непереносимість фруктози. Сімейні варіанти синдрому успадковуються аутосомно-рецесивно, аутосомно-домінантно або зчепленно з X- хромосомою[3] .
Тип успадкування — аутосомно- рецесивний, виділена також аутосомно -домінантна форма з локалізацією гена на хромосомі 15q15.3 . Експресивність мутантного гена в гомозиготному стані значно варіює. Зустрічаються спорадичні випадки, обумовлені свіжою мутацією. Вважається, що в основі хвороби лежать генетично зумовлені дефекти ферментативного фосфорилювання в ниркових канальцях (комбінована тубулопатія), дефіцит ферментів 2-го і 3-го комплексів дихального ланцюга — сукцинатдегідрогеназного і цитохромоксидазного. Вчені відносять захворювання до розряду мітохондріальних хвороб.
Патогенез
Патологічні зміни являють собою один з варіантів вторинного гіперпаратиреозу. Основна ланка патогенезу — мітохондріальний ферментний дефект в циклі Кребса, ферментна тубулопатія, що характеризується порушенням реабсорбції глюкози, амінокислот, фосфатів і бікарбонатів в канальцях нирок.[2] Втрата амінокислот і бікарбонату сприяє розвитку метаболічного ацидозу, на тлі якого посилюється резорбція кісткової тканини і знижується реабсорбція калію і кальцію в канальцях нирок, що призводить до розвитку гіпокаліємії і гіперкальціурії. Втрата фосфору веде до розвитку рахіту, а у дітей старшого віку і дорослих — до остеомаляції.[3]
Таким чином, мітохондріальний ферментний дефект в циклі Кребса веде до порушення процесів енергозабезпечення, реабсорбції фосфатів , глюкози і амінокислот в ниркових канальцях і підвищеної їх екскреції з сечею — порушується кислотно — основний стан, а метаболічний ацидоз і брак фосфатів сприяють руйнуванню кісткової тканини по типу рахітоподібних змін скелета і остеомаляції .
Клінічні ознаки
Перші ознаки захворювання з'являються в другому півріччі життя — діти мляві, гіпотрофічні, апетит різко знижується, спостерігаються блювання, субфебрилітет, гіпотонія, спрага, поліурія, дегідратація[2] . Розгорнутий симптомокомплекс формується до другого року життя. Якщо захворювання маніфестує в 5-6 років, то першими ознаками є симптоми остеомаляції, деформація кісток і гіпокаліємічні паралічі[3]. З другого року життя виявляють відставання фізичного і інтелектуального розвитку, відбувається генералізована декальцифікація, що проявляється кістковими деформаціями ніг (вальгусні або варусні), грудної клітини, передпліч і плечових кісток, зниженням м'язового тонусу. Рентгенологічно виявляють деформацію кісток, хребетного стовпа[2], переломи, системний остеопороз різного ступеня вираженості, витончення коркового шару трубчастих кісток, розпушення зон росту, відставання темпів росту кісткової тканини від паспортного віку дитини. Кістки стають ламкими.
При лабораторному обстеженні виявляють нормо або гіпокальціємію, гіпофосфатемію, підвищений рівень лужної фосфатази. У результаті зниження реабсорбції бікарбонатів в канальцях нирок спостерігається гіперхлоремічний ацидоз на тлі надлишку паратгормону і нормо- або гіпокальціємії. У біохімічному аналізі сечі виявляють аміноацидурію, глюкозурію (при нормальних рівнях глікемії), натрійурію, гіпокальційурію на тлі гіперфосфатемії[2] .
Залежно від тяжкості клінічних проявів і метаболічних розладів виділяють два клініко- біохімічних варіанти хвороби:
- Перший характеризується значною затримкою фізичного розвитку, важким перебігом захворювання з вираженими кістковими деформаціями і нерідко переломами кісток, різкою гіпокальціємією (1.6-1.8 ммоль/л), зниженням абсорбції кальцію в кишечнику.
- При другому варіанті відзначають помірну затримку фізичного розвитку, легкий перебіг з незначними кістковими деформаціями, нормокальціємією і нормальним засвоєнням кальцію в кишечнику.
Біохімічні порушення
- зниження рівня кальцію в крові
- зниження рівня фосфору в крові
- підвищення рівня лужної фосфатази
- розвиток метаболічного ацидозу (рН: 7,35 … 7,25; ВЕ : −10 … −12 ммоль/л) за рахунок дефекту реабсорбції бікарбонатів у проксимальних канальцях
- нормальна екскреція кальцію з сечею
- підвищення кліренсу фосфатів сечі, всмоктування фосфатів у кишечнику не порушується
- розвиток глюкозурії (20-30 г/л і вище)
- розвиток генералізованої гіпераміноацідурії
- порушення функцій амоніоацидогенезу — зниження титраційної кислотності, підвищення рН сечі більше 6.0
- розвиток гіпокаліємії
Результатом захворювання є розвиток хронічної ниркової недостатності.[2]
Диференціальний діагноз
Диференціальну діагностику синдрому де Тоні — Дебре — Фанконі проводять з рахітом і рахітоподібними захворюваннями у дітей.[2] Також диференціюють із вторинним синдромом, що розвивається на тлі інших спадкових і набутих захворюваннях:
- синдромі Лоу
- ювенільному нефронофтизі
- цистинозі
- тирозинемії
- галактоземії
- глікогенозах
- спадковій непереносимості фруктози
- Rod — cone дистрофії
- гепатобіліарній дистрофії
- мієломній хворобі
- амілоїдозі
- синдромі Шегрена
- нефротичному синдромі
- нирковій трансплантації
- гіперпаратиреозі, ураженні нирок солями важких металів
- отруєнні лікарськими речовинами, у тому числі вітаміном D, лізолом і т. д.
Лікування
Основні принципи — корекція електролітних порушень, зрушень у кислотно-лужній рівновазі, усунення дефіциту калію і бікарбонатів. Збільшують споживання фосфору з їжею, обмежують споживання продуктів, що включають сірковмісні амінокислоти, призначають великі дози вітаміну D. Для лікування цистинозу з метою зниження накопичення цистину в тканинах і проксимальних ниркових канальцях застосовують меркаптамін. Призначають препарати кальцію і вітаміну D, при хронічній нирковій недостатності проводиться гемодіаліз.[2]
Примітки
- Whonamedit?- A dictionary of medical eponyms. De Toni-Debré-Fanconi syndrome
- Малая энциклопедия врача-эндокринолога / Под ред. А. С. Ефимова. — 1-е изд. — К.: Медкнига, ДСГ Лтд, Киев, 2007. — С. 340. — 360 с. — («Библиотечка практикующего врача»). — 5000 экз. — ISBN 966-7013-23-5
- Симптомы и синдромы в эндокринологии / Под ред. Ю. И. Караченцева. — 1-е изд. — Х.: ООО «С. А. М.», Харьков, 2006. — С. 165—166. — 227 с. — (Справочное пособие). — 1000 экз. — ISBN 978-966-8591-14-3
- G. de Toni: Remarks on the relation between renal rickets (renal dwarfism) and renal diabetes. Acta pædiatrica, Stockholm, 1933, 16: 479.
- R. Debré, J. Marie, F. Cleret, Messimy: Rachitisme tardif coexistant avec une néphrite chronique et une glycosurie. Arch Méd Enf Paris, 1934, 37: 597.
- G. Fanconi: Der frühinfantile nephrotisch-glykosurische Zwergwuchs mit Hypophosphatämischer Rachitis. Jahrbuch für Kinderheilkunde, Berlin, 1936, 147: 299.