Хвороба Віллебранда
Хвороба Віллебранда (англ. Von Willebrand disease) — спадкове захворювання крові, що характеризується виникненням епізодичних спонтанних кровотеч, які схожі з кровотечами при гемофілії. Захворювання успадковується за принципом аутосомного домінування і пов'язане з дефіцитом фактора фон Віллебранда, який бере участь в адгезії тромбоцитів на коллагені і захищає VIII фактор від протеолізу. При дефіциті фактора фон Віллебранда, VIII фактор піддається протеолізу і його вміст у плазмі знижується. Крім того при хворобі Віллебранда знижується вміст серотоніну і розвивається патологічна дилятація судин та підвищення їх проникності. При хворобі Віллебранда спостерігаються найдовші кровотечі через те, що у хворих порушені всі три ланки гемостазу.
| Хвороба Віллебранда | |
|---|---|
| Спеціальність | гематологія |
| Симптоми | кровотеча, носова кровотеча, крововилив, Геморагічні діатези, гематома, Гемартроз, шлунково-кишкова кровотеча, геморагічний інсульт і Субарахноїдальний крововилив |
| Причини | Фактор фон Віллебранда |
| Препарати | Десмопресин[1] і Antihemophilic Factord[1] |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | 3B12 |
| МКХ-10 | D68.0 |
| OMIM | 193400 |
| DiseasesDB | 14007 |
| eMedicine | ped/2419 |
| MeSH | D014842 |
Є три форми хвороби Віллебранда: успадкований, набутий та псевдо (або тромбоцитарний) тип. Виокремлюють три типи спадкової хвороби Віллебранда: ХВ Тип I, ХВ Тип II, і ХВ III. Для Типу ІІ ХВ існують чотири різні підтипи. Тромбоцитарний тип ХВ також є спадковим захворювання. ХВ I типу найпоширеніша, і протікає, як правило, безсимптомно або проявляється легкими симптомами, такими як носові кровотечі. Існують різні фактори, які впливають на прояви і тяжкість симптомів ХВ, такі як група крові.
Причина кровотеч — порушення згортання крові через недостатню активність фактора Віллебранда. Поширеність хвороби Віллебранда становить 1 на 800—1000.
Хвороба Віллебранда названа на честь Еріка Адольфа фон Віллебранда, фінського педіатра, що вперше описав захворювання в 1926 році.
Синоніми
Ангіогемофілія; атромбопенічний пурпур; атромбоцитопенічний пурпур; геморагічна капіляропатія; конституціональна тромбопатія, конституціональна тромбопатія фон Віллебранда-Юргенса; спадкова псевдогемофілія; синдром Юргенса; судинна гемофілія, псевдогемофілія.
Класифікація
Є три тип хвороби Віллебранда[2]:
- успадкований (спадковий):
- Тип I
- Тип II:
- A
- B
- M
- N
- Тип III
- набутий
- псевдо (або тромбоцитарний)
- 1-й тип обумовлений частковим кількісним дефіцитом фактора Віллебранда. При цьому його мультимірна структура збережена. Наявне зниження прокоагулянтної активності фактора VIII, агрегації тромбоцитів, індукованої рістоцетіном, рістоцетінкофакторної активності, антигену фактора Віллебранда. Частота даної форми становить від 75 % до 80 % всіх випадків хвороби Віллебранда. Спадкування аутосомно-домінантне.
- 2-й тип обумовлений якісними змінами фактора Віллебранда, пов'язаний з порушенням формування мультимерів і підрозділяється на підтипи: 2A, 2B, 2M, 2N. Спадкування хвороби Віллебранда 2-го типу аутосомно-домінантне, за винятком підтипу 2N, де воно рецессивне. Частота появи даних форм становить від 5 % до 15 % всіх випадків хвороби Віллебранда.
- Фенотип підтипу 2A є результатом порушення двох різних механізмів: дефекту синтезу високомолекулярних мультимерів та підвищення протеолізу фактора Віллебранда. При підтипі 2B відзначається підвищену спорідненість фактора Віллебранда до рецептора на мембрані тромбоцитів глікопротеїну Ib.
- Підтип 2В.Це дефект «посилення функції». Здатність якісно дефектного фактора фон Віллебранда прив'язуватись до глікопротеїну1 (GP1) рецептора на мембрані тромбоцитів аномально підвищена, що призводить до його спонтанного зв'язування з тромбоцитами і подальшого швидкого кліренсу з пов'язаних тромбоцитів і великих мультимерів фактора Віллебранда. Може виникнути тромбоцитопенія, а великі мультимери Віллебранда зникнути з циркуляції.
- Підтип 2M характеризується порушенням зв'язку фактора Віллебранда з рецептором глікопротеїном Ib на мембрані тромбоцитів.
- Підтип 2N характеризується нормальним рівнем фактора Віллебранда і низькою прокоагулянтною активністю, що зумовлено порушенням зв'язку фактора VIII і фактора Віллебранда.
- 3-й тип — найбільш важка форма з повним дефіцитом фактора Віллебранда. Ця форма характеризується відсутністю фактора Віллебранда в плазмі, тромбоцитах і судинній стінці. Рівень фактора VIII нижче 10 %. Спадкування — аутосомно-рецесивне. Захворювання проявляється у гомозигот з однаковими дефектними алелями або у подвійних гетерозигот з двома різними дефектними алелями. У пацієнтів з 3-м типом мається ймовірність появи аллоантитіл до фактору Віллебранда. Частота народження з захворюванням 3-го типу хвороби Віллебранда менше 5 %.
Крім того, існує тромбоцитарний тип хвороби Віллебранда, який обумовлений мутацією в гені тромбоцитарного рецептора глікопротеїну Ib, внаслідок якої підвищується чутливість даного рецептора до високомолекулярних мультимерів фактора Віллебранда. Фенотип аналогічний підтипу 2B.
- Набутий синдром Віллебранда визначається у пацієнтів з аутоімунними, лімфопроліферативними захворюваннями, обумовлений появою інгібітору проти фактора Віллебранда, а також якісними аномаліями фактора VIII у зв'язку з адсорбцією високомолекулярних мультимерів патологічними білками.
Набута хвороба Віллебранда
Набута хвороба Віллебранда може виникнути у хворих з аутоантитілами. У цьому випадку функція фактора Віллебранда не інгібована, але комплекс фактор Віллебранда — антитіло швидко виводиться з кровотоку.
Ще одна Форма ХВ виникає у пацієнтів з аортальним стенозом, що призводить до шлунково—кишкових кровотеч (синдром Хейда — англ. Heyde's syndrome). У 2003 р. з'ясувалося, що пацієнти з набутою хворобою Віллебранда і аортальним стенозом, які перенесли протезування клапанів, потребували корекції аномалій гемостазу. Але ці аномалії могли проявлятися повторно за 6 місяців, якщо протез клапана погано підходив пацієнту[3]. Крім того, з'являється схильність до кровотеч у людей з імплантатом лівого шлуночка (Left Ventricular Assist Device — LVAD), насос, що качає кров з лівого шлуночка серця в аорту[4]. В обох випадках спостерігається руйнування великих мультимерів фактора Віллебранда, внаслідок механічного тиску та навантажень.
Тромбоцитоз є ще однією причиною набутої хвороби Віллебранда, у зв'язку з секвестрацією фактора Віллебранда через адгезію величезного числа тромбоцитів.
Набута хвороба Віллебранда також була описана при наступних порушеннях: пухлини, гіпотиреоз і мезенхімальні дисплазії Вільмса.
Патогенез
ФВ найбільш активний в умовах високого кровотоку і напруги. Дефіцит фактора Віллебранда, проявляється найперше в органах з великою кількістю дрібних судин, таких як шкіра, шлунково — кишковий тракт і матка. У ангіодисплазії, форма телеангіоектазії товстої кишки, напруга набагато вище, ніж у середньому в капілярах і тому ризик кровотечі збільшується.
При тяжких випадках типу 1 ХВ, всі генетичні зміни відбуваються в гені, що кодує функціональні характеристики фактора Віллебранда і характеризуються високою пенетрантністю. При легких формах типу 1 ХВ широкий спектр молекулярної патології може виникати на додаток до поліморфізму гена. Група крові людини (система АВО) також може впливати на перебіг захворювання, визначаючи її симптоми і тяжкість впливу на організм. Як правило, у осіб, з I(O) групою крові, середній рівень фактора Віллебранда нижче ніж у людей з іншими групами крові. Саме тому при дослідженні рівня фактора Віллебранда за допомогою системи ABO слід пам'ятати про вищевказану особливість. Адже відомі такі випадки, коли здоровим людям з I (О) групою крові діагностували I тип ХВ, в той час, як лдей з IV (АВ) групою крові, у яких були присутні певні генетичні порушення вважали цілком здоровими, у зв'язку з підвищеним в цій групі крові природним рівнем ФВ[5].
Генетика
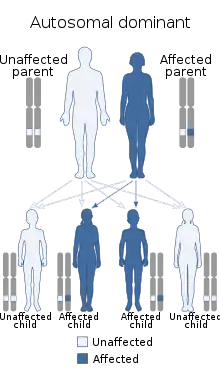
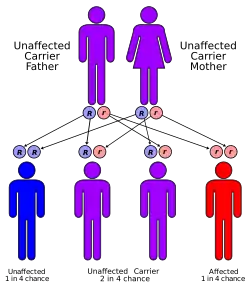
Ген фактора Віллебранда знаходиться на дванадцятій хромосомі(12p13.2). Він має 52 екзони, що охоплюють 178kbp. Типи 1 і 2 успадковуються як аутосомно-домінантна ознака, а тип 3 має аутосомно-рецесивне успадкування. Іноді тип 2 також успадковується рецессивно.
Епідеміологічні особливості
Поширеність ХВ становить близько 1 в 100 осіб[6]. Однак більшість з цих людей не мають ніяких симптомів. Поширеність клінічно значущих випадків становить 1 на 10 000. Оскільки більшість форм досить м'які, вони виявляються частіше у жінок, чия схильність до кровотеч проявляється під час менструації. Ці прояви можуть бути більш важкими та вираженими у людей з групою крові І(О).
Клінічні ознаки
Найбільш характерними і специфічними симптомами при хворобі Віллебранда є кровотечі з слизових порожнини рота, носа, внутрішніх органів. Симптоми кровоточивості варіюють від помірно виражених до вкрай тяжких, перебігають переважно по мікроциркуляторному типу. У пацієнтів з різким дефіцитом фактора VIII спостерігаються рясні і тривалі кровотечі (носові, ясенні, маткові), також крововиливи в м'язи і суглоби. Крім того, можуть виникати тривалі кровотечі при травмах, видаленні зубів, операціях.
У дитячому віці часто бувають кровотечі з слизових оболонок порожнини рота, носові кровотечі, синці на шкірі. Тяжчий перебіг геморагічного діатезу відзначається під час, або незабаром після перенесених інфекційних захворювань. Найбільш імовірним пусковим механізмом кровотечі на тлі інфекційної хвороби є порушення проникності судин. Внаслідок цього з'являються мимовільні кровотечі діапедезного типу.
Гематоми — крововиливи в підшкірну клітковину і м'язові тканини спостерігаються переважно після травм у хворих з тяжкими формами захворювання.
При хворобі Віллебранда геморагічний синдром проявляється не завжди, періоди загострення чергуються з періодами повної або майже повної відсутності геморагій. У деяких пацієнтів хвороба Віллебранда може поєднуватися з ознаками мезенхімальної дисплазії: підвищеною еластичністю шкіри, слабкістю, підвищеною рухливістю суглобів, пролапсом стулок клапанів серця.
Аутосомний тип спадкування обумовлює однакову частоту виникнення хвороби Віллебранда у пацієнтів обох статей. У жінок внаслідок особливостей фізіологічної будови організму, пов'язаних з репродуктивною функцією, спостерігається більш частий прояв геморагічних симптомів. Близько 65 % жінок з хворобою Віллебранда страждають менорагіями. Рецидивуючі маткові кровотечі, що тривають більше 10 днів, супроводжуються постгеморрагічною анемією.
Шлунково — кишкові кровотечі у пацієнтів з хворобою Віллебранда не є основною формою кровоточивості. Вони можуть бути спричинені вживанням лікарських препаратів, що впливають на агрегацію тромбоцитів (ацетилсаліцилова кислота та інші нестероїдні протизапальні засоби). Крім того, джерелами кровотеч є латентні виразки шлунка та дванадцятипалої кишки, також ерозивні гастрити, гемороїдальні вузли.
У пацієнтів з хворобою Віллебранда можуть бути тривалі кровотечі при операціях, у жінок — під час пологів. Пологи у жінок з хворобою Віллебранда пов'язані з ризиком виникнення значної крововтрати. У більшості пацієнтів з середньою і легкими формами захворювання під час вагітності рівень фактора VIII підвищується в 2-3 рази і сягає нормальних значень, проте в післяпологовому періоді повертається до вихідного рівня.
Гемартроз — найбільш рідкісний прояв хвороби Віллебранда, характерний для захворювання 3-го типу. Гострий гемартроз супроводжується больовим синдромом, обумовленим підвищенням внутрішньосуглобового тиску. Суглоб збільшений в об'ємі, шкіра над ним гіперемована і гаряча на дотик. Якщо гемартроз виник після травми, потрібно виключити додаткові пошкодження (внутрішньосуглобовий перелом, відрив відростка, утиск тканин). Рецидивуючі гемартрози спричинюють хронічний синовіт. На стадії синовіту синовіальна оболонка гіпертрофується і стає основним джерелом крововиливу в суглоб. При гострому синовіті гемартрози можуть рецидивувати, незважаючи на трансфузії фактора згортання VIII, що обумовлено запальним процесом в синовіальній оболонці. При хронічному синовіті больовий синдром може бути відсутнім, оскільки зруйнована капсула суглоба.
На відміну від гемофілії, при хворобі Віллебранда подальшого прогресування патологічного процесу та розвитку деформуючого остеоартрозу, як правило, не спостерігається.
Крововиливи в головний і спинний мозок і їх оболони при хворобі Віллебранда виникають у зв'язку з травмою. В окремих випадках причиною таких крововиливів може бути гіпертонічний криз або вживання препаратів, що значно порушують гемостатичну функцію тромбоцитів (ацетилсаліцилова кислота, бутадіон тощо).
Враховуючи аутосомно-домінантний тип успадкування, генетичний ризик для потомства становить 50 % незалежно від статі плоду.
Діагностика
Діагностувати хворобу Віллебранда в легкій формі її перебігу практично неможливо, оскільки кровотечі при ній виникають не часто. Людина може помітити тільки сильну кровотечу при тяжкій травмі, або після хірургічного втручання стоматолога.
Якщо в особи є підозра на наявність хвороби Віллебранда, то для точної діагностики необхідно провести кількісне і якісне дослідження плазми крові пацієнта на наявність фактора Віллебранда. Цей процес здійснюється шляхом вимірювання рівня фактора Віллебранда (антигенів цього фактора), що містяться в досліджуваному зразку крові. Для перевірки функціональності цього фактора зазвичай визначають рівень зв'язування фактора Віллебранда з глікопротеїном (GP) Ib, його зв'язок з колагеном. Іншими методами перевірки активності ФВ може бути визначення активності кофактору рістоцетіну (RiCof) або швидкість аглютинації тромбоцитів, після введення рістоцетіну (ristocetin induced platelet agglutination (RIPA)).
Крім того, може бути здійснена перевірка рівня фактора VIII, адже фактор Віллебранда перешкоджає швидкому руйнуванню фактора VIII в крові. Тобто, дефіцит фактора Віллебранда може привести до різкого зниження рівня фактора VIII. Однак, навіть нормальний рівень цих коагуляційних факторів не виключає наявність захворювання, особливо якщо мова йде про 2 тип ХВ. У такому випадку хвороба може бути виявлена тільки шляхом дослідження взаємодії тромбоцитів з субендотеліальним шаром у кровотоці (фактор активації тромбоцитів PAF), що проводиться тільки у вузькоспеціалізованих лабораторіях і зазвичай не здійснюється в більшості звичайних медичних лабораторій[7][8]. Аналіз агрегації тромбоцитів, як правило, показує ненормальну реакцію на введення рістоцетіну, проте реакція на інші антагоністи буде нормальною. Тип 2N хвороби Віллебранда може бути діагностований тільки після проведення аналізу активності фактора VIII . Виявлення ХВ ускладнюється ще й тим, що фактор Віллебранда — це гострофазовий білок, тобто його рівень в організмі різко підвищується під час вагітності, при стресі та інфекційних захворюваннях.
Лікування
Пацієнтам з хворобою Віллебранда, як правило, ніякого регулярного лікування не потрібно, хоча ризик кровотечі у них завжди підвищений. Для жінок з тяжкими менструальними кровотечами, можуть бути рекомендовані комбіновані оральні протизаплідні, які, як правило, знижують кровотечі, скорочують тривалість і частоту менструації. Іноді, хворим на ХВ, яким планується здійснювати хірургічну операцію проводять профілактичне лікування. У такому випадку, людям зазвичай вводять препарати, основою яких є концентрати фактора VIII в комплексі з фактором Віллебранда (плазматичні антигемофільні фактори). Якщо перебіг захворювання є помірним, то профілактика кровотеч може здійснюватися за допомогою десмопресину (1-desamino-8-D-аргінін вазопресину[джерело?], англ. DDAVP, десмопресин ацетат), який підвищує рівень фактора Віллебранда в плазмі пацієнта.
Історичні факти
У 1924 році 5 — річна дівчина, яка жила на Аландських островах була доставлена в лікарню в Гельсінкі, Фінляндія, де вона була помічена доктором Еріком фон Віллебрандом. Він, в кінцевому рахунку, обстежив 66 членів її сім'ї та повідомив в 1926 році, що він відкрив раніше не описане порушення згортання крові, що відрізняється від гемофілії. Доктор фон Віллебранд визначив аутосомне успадкування і відзначив, що симптоми кровотечі були більш виражені у дітей і жінок дітородного віку. Таким чином, він заявив, що пацієнти з цим синдромом мали шкірно — слизові кровотечі, нормальний час згортання, аутосомне успадкування замість пов'язаного з Х — хромосомою і тривалу кровотечу за методом Дюка. Згодом він виявив, що переливання крові були дієвими не тільки при корекції анемії, але і для зупинки кровотеч[9].
У 1950 році стало ясно, що в цих осіб був знижений «фактор плазми», антигемофільний фактор (фактор VIII). З цього часу, фактор, що спричинює тривалі кровотечі, почали називати «фактором Віллебранда» на честь доктора Еріка фон Віллебранда.
У 1980-х, молекулярні та клітинні дослідження відрізнили гемофілію А і хворобу Віллебранда точніше. Особи, які хворіли на ХВ, мали нормальний ген фактора VIII на Х-хромосомі, але в них був аномальний ген фактора Віллебранда на 12 хромосомі.
Див. також
Примітки
- NDF-RT
- Sadler JE (1994). A revised classification of von Willebrand disease. For the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb. Haemost. 71 (4): 520–5. PMID 8052974.
- Vincentelli A, Susen S, Le Tourneau T, et al. (July 2003). Acquired von Willebrand syndrome in aortic stenosis. N. Engl. J. Med. 349 (4): 343–9. PMID 12878741. doi:10.1056/NEJMoa022831.
- Uriel, N; Pak SW, Jorde UP, Jude B, Susen S, Vincentelli A, Ennezat PV, Cappleman S, Naka Y, Mancini D (5 жовтня 2010). Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol. 56(15) (15): 1207–13. PMID 20598466. doi:10.1016/j.jacc.2010.05.016.
- Gill, JC; Endres-Brooks J, Bauer PJ, Marks WJ, Montgomery RR (1987). The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood 69 (6): 1691–5. PMID 3495304. Архів оригіналу за 29 січня 2005.
- Molecular basis of von Willebrand disease and its clinical implications. Haematologica 89 (9): 1036. 1 вересня 2004. PMID 15377463.
- Favaloro EJ, Bonar R, Kershaw G, et al. (July 2006). Reducing errors in identification of von Willebrand disease: the experience of the Royal College of Pathologists of Australasia quality assurance program. Semin. Thromb. Hemost. 32 (5): 505–13. PMID 16862524. doi:10.1055/s-2006-947865.
- Chandler WL, Peerschke EI, Castellone DD, Meijer P (June 2011). Von Willebrand factor assay proficiency testing. The North American Specialized Coagulation Laboratory Association experience. Am. J. Clin. Pathol. 135 (6): 862–9. PMID 21571959. doi:10.1309/AJCPH5JK4ONENPAE.
- Von Willebrand, EA (May 1999). Hereditary pseudohaemophilia. Haemophilia : the official journal of the World Federation of Hemophilia 5 (3): 223–31; discussion 222. PMID 10444294. doi:10.1046/j.1365-2516.1999.00302.x.