Панкреатична нейроендокринна пухлина
Панкреатична нейроендокринна пухлина (ПанНЕП, англ. PanNET, PNET) — відносно рідкісне новоутворення підшлункової залози, що походить з нейроендокринних клітин острівця підшлункової залози та класифікується як нейроендокринна пухлина. ПанНЕП може бути функціонуючою або нефункціонуючою в залежності від гормональної активності. ПанНЕП часто демонструє інвазію та метастазування, але відрізнити злоякісно трансформовану пухлину від доброякісної ПанНЕП при аналізі патогістологічних слайдів неможливо.[3]
| Панкреатична нейроендокринна пухлина | |
|---|---|
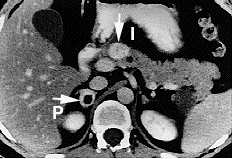 КТ знімок: в голівці підшлункової залози визначається новоутворення - ПанНЕП (стрілка вгорі). Також визначається новоутворення правого наднирника - феохромоцитома. КТ знімок: в голівці підшлункової залози визначається новоутворення - ПанНЕП (стрілка вгорі). Також визначається новоутворення правого наднирника - феохромоцитома. | |
| Спеціальність | онкологія |
| Препарати | Дакарбазин[1], еверолімус[2] і бевацизумаб[2] |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | XH5XB7 |
| МКХ-10 | C25.4 |
| МКХ-О | 8150/3 і D3A.8 |
| OMIM | 260350 |
| eMedicine | med/ |
| MeSH | D010190 |
| | |
Етіологія
Переважна більшість ПанНЕП виникає спорадично, але певна пропорція пухлин діагностується як складова синдрому множинної ендокринної неоплазії типу 1 або хвороби фон Гіппеля-Ліндау.
Патологічна анатомія
ПанНЕП може характеризуватися трабекулярним, альвеолярним, солідним, псевдозалозистим, ангіоматозним або ациноподібним типом будови. При електронній мікроскопії клітини ПанНЕП містять нейросекреторні гранули та демонструють прозорі везикули, що відповідають нейро-секреторним везикулам.
Генетичний профайл
Молекулярні та генетичні дослідження ПанНЕП виявили декілька мутацій, визначення яких може потенційно бути застосовано з діагностичною метою.[4][5] Були виявлені наступні мутовані гени, що показали інші типи SNP у порівнянні з раком підшлункової залози:
- MEN1, ATRX, DAXX[4][6][7]
- Мутації в генах сигнального шляху mTOR: TSC2, PTEN, PIK3CA.[4] Визначення даних мутацій має клінічне значення, оскільки існують (хоча і в режимі клінічних випробовувань) препарати інгібітори mTOR
- Мутації в генах ATRX та DAXX визначаються приблизно у 40% ПанНЕП.[4] Ці гени кодують однойменні білки, що беруть участь у процесах ремоделювання хроматину теломер.
Клінічна картина
Нефункціонуючі ПанНЕП
Нефункціонуючі ПанНЕП не мають специфічних симптомів, через що часто діагностуються на пізніх стадіях у зв'язку з розмірами пухлини або наявністю метастазів (локальних чи віддалених). Біль в животі спостерігається у більшості хворих, до 35% випадків демонструють втрату ваги, анорексія та нудота до 45% випадків. Також можуть спостерігатись жовтяниця в 20-50% пацієнтів, пальпаторне визначення пухлини в 10-40% пацієнтів.[8]
Функціонуючі ПанНЕП
До функціонуючих ПанНЕП належать пухлини підшлункової залози, що секретують гормони. В залежності від типу секретованого гормону буде відповідна клінічна картина. Серед цих пухлин найчастіше діагностується інсулінома (17%), гастринома (15%). Рідше визначаються ВІПома (2%), глюкагонома (1%), карциноїд (1%), соматостатинома (1%), а також адренокортикотропін-секретуючі пухлини (АКТГ-ома), гормон рилізинг-фактор секретуючі пухлини (ГРФоми), кальцитонін продукуючі пухлини, що визначаються дуже рідко.[9]
Діагностика
Cцинтіграфія
Соматостатин-рецепторна сцинтіграфія з використанням радіоактивного аналогу соматостатину октреотиду є високоінформативним діагностичним засобом для визначення ПанНЕП. З новітніх засобів високий потенціал має ПЕТ з використанням Ga-DOTATOC для візуалізації рецепторів соматостатину.
Ультразвукове дослідження
УЗД відносно чутливий метод. ПанНЕП при УЗД визначається як гіпоехогенне утворення.
Комп'ютерна томографія
При підозрі на ПанНЕП застосовують КТ без контрастування.
Магнітно-резонансна томографія
МРТ використовують при підозрі на ПанНЕП та віддіалені метастази. За допомогою МРТ можуть бути визначені метастази < 1 см.
Лікування
Пацієнтам з ПанНЕП застосовують загальні підходи до лікування нейроендокринних пухлин. Функціонуючі пухлини лікують за допомогою аналогів соматостатину. При ідентифікації пухлини проводять її видалення хірургічним шляхом.
Посилання
- NDF-RT
- Drug Indications Extracted from FAERS — doi:10.5281/ZENODO.1435999
- Н.Б. Губергріц, П.Г. Фоменко, Г.М. Лукашевич, О.О. Голубова (2011). Ендокринні пухлини підшлункової залози: що треба знати лікарю. Газета «Новости медицины и фармации» Гастроэнтерология 390.
- Jiao, Y.; Shi, C.; Edil, B. H.; De Wilde, R. F.; Klimstra, D. S.; Maitra, A.; Schulick, R. D.; Tang, L. H.; Wolfgang, C. L.; Choti, M. A.; Velculescu, V. E.; Diaz Jr, L. A.; Vogelstein, B.; Kinzler, K. W.; Hruban, R. H.; Papadopoulos, N. (2011). DAXX/ATRX, MEN1, and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science 331 (6021): 1199–1203. PMID 21252315. doi:10.1126/science.1200609.
- McKenna, L. R.; Edil, B. H. (2014). Update on pancreatic neuroendocrine tumors. Gland surgery 3 (4): 258–275. PMID 25493258. doi:10.3978/j.issn.2227-684X.2014.06.03.
- Jones, S.; Zhang, X.; Parsons, D. W.; Lin, J. C. -H.; Leary, R. J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; Hong, S. -M.; Fu, B.; Lin, M. -T.; Calhoun, E. S.; Kamiyama, M.; Walter, K.; Nikolskaya, T.; Nikolsky, Y.; Hartigan, J.; Smith, D. R.; Hidalgo, M.; Leach, S. D.; Klein, A. P.; Jaffee, E. M.; Goggins, M.; Maitra, A.; Iacobuzio-Donahue, C.; Eshleman, J. R.; Kern, S. E.; Hruban, R. H. (2008). Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 321 (5897): 1801–1806. PMID 18772397. doi:10.1126/science.1164368.
- PMID 19077451 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - Falconi M, Plockinger U, Kwekkeboom DJ, Manfredi R, Korner M, Kvols L, Pape UF, Ricke J, Goretzki PE, Wildi S, Steinmuller T, Oberg K, Scoazec JY; Frascati Consensus Conference; European Neuroendocrine Tumor Society. (2006). Well-Differentiated Pancreatic Nonfunctioning Tumors/Carcinoma. Neuroendocrinology 84 (3): 196–211. PMID 17312380. doi:10.1159/000098012.
- O'Toole D, Salazar R, Falconi M, Kaltsas G, Couvelard A, de Herder WW, Hyrdel R, Nikou G, Krenning E, Vullierme MP, Caplin M, Jensen R, Eriksson B; Frascati Consensus Conference; European Neuroendocrine Tumor Society. (2006). Rare functioning pancreatic endocrine tumors. Neuroendocrinology 84 (3): 189–195. PMID 17312379. doi:10.1159/000098011.