Синдром Марфана
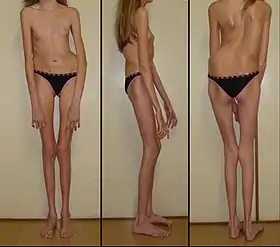
Синдром Марфана — генетичне порушення розвитку сполучної тканини.[5] Ступінь порушення варіює. Хворі на синдром Марфана зазвичай є високими, худими, з довгими руками, ногами, пальцями і ступнями, також для них характерні надзвичайно гнучкі суглоби і сколіоз.[5] Найбільш серйозні ускладнення зустрічаються з боку аорти і мітрального клапана — у хворих часто діагностують аневризму аорти та недостатність мітрального клапана через пролапс його задньої стулки.[5][6] Інші органи, в яких можуть виникати зміни включають легені, очі, кістки та хребет.[5]
| Синдром Марфана | |
|---|---|
 | |
| Спеціальність | медична генетика |
| Симптоми | доліхостеномеліяd[1], Плоскостопість[1], пролапс мітрального клапануd[2], сколіоз[3], pectus excavatumd[3], pectus carinatumd[3], аневризма аорти[3], диссекція аорти[3] і вивих кришталикаd[4] |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | LD28.01 |
| МКХ-10 | Q87.4 |
| OMIM | 154700 |
| DiseasesDB | 7845 |
| MedlinePlus | 000418 |
| eMedicine | ped/1372 |
| MeSH | D008382 |
| | |
Синдром Марфана — аутосомно-домінантне захворювання. Приблизно у 75 % випадків воно є успадковане, решта 25 % спричинені випадковими мутаціями.[5] Мутація зазвичай виникає у гені, який містить інформацію про синтез фібриліну-1, що викликає порушення структури сполучної тканини.[5] Діагноз про наявність синдрому Марфана у хворого часто базується на використанні критерій Гента.
Немає причинної медичної терапії від синдрому Марфана. Більшість хворих живуть звичайно, використовуючи ситуативне лікування.[5] Воно часто включає використання бета блокаторів, таких як пропранолол або, у разі наявної сенсибілізації до нього, блокаторів кальцієвих каналів та ACE інгібіторів.[7][8] Хірургічне втручання може бути необхідне для корекції аневризми аорти та мітрального клапана.[8] До загальних рекомендацій для хворих на синдром Марфана відносять уникнення виконання важких фізичних вправ.[7]
Зустрічається рідко — 1:3.000-10.000 від загальної кількості новонароджених мають синдром Марфана.[7][9] Зустрічається однаково часто як серед чоловіків так і серед жінок, як і всі автосомно-домінантні захворювання.[7] Частота виникнення не пов'язана з расою і з регіоном.[9] Синдром названий в честь Антуана Марфана, французького педіатра, який вперше описав захворювання у 1896.[10][11]
Історія
Синдром Марфана був названий на честь Антуана Марфана,[10] французького педіатра, хто вперше описав це стан у 1896 році, після того, як помітив типові ознаки у 5-річної дівчинки.[11][12] Ген, пов'язаний із захворюванням був уперше виділений Франчезко Рамірезом у Медичному центр Маунт Синай (Mount Sinai Medical Center), міста Нью-Йорк у 1991.[13]
Епідеміологічні особливості
Досліджено, що на синдром Марфана жінки і чоловіки хворіють однаково часто,[14] і мутація не залежить від етнічних і географічних біомів.[9] Частота хворих становить 1:3.000-10.000.[7][9]
Патогенез
Синдром Марфана спричиняється мутаціями FBN1 гена 15 хромосоми,[15] що кодує фібрилін-1 — глікопротеїновий компонент екстрацелюлярного матриксу. Фібрилін-1 є необхідний для правильного формування екстрацелюлярного матриксу, а саме біогенезу і підтримання еластичних волокон. Екстрацелюлярний матрикс є критично необхідний для структурної інтеграції сполучної тканини, а також слугує резервуаром для факторів росту.[16] Еластичні волокна існують у кожному куточку людського тіла, проте найважливішу функцію виконують в аорті, сухожилках і війчастому пояску ока; тому ці зони і є найбільш вразливі.
Трансгенетичні миші-носії єдиної копії мутантного фібриліну-1, мутації схожої на ту, яка виникає в людському гені, спричинює у мишей розвиток синдрому Марфана. Миші з цим дефектним геном мають схожі прояви з людськими. Окрім цього, зниження вмісту нормального фібриліну-1 породжує Марфан-пов'язані захворювання у мишей.
Трансформуючий фактор росту бета (TGF-β) відіграє важливу роль у розвитку синдрому Марфана. Фібрилін-1 безпосередньо пов'язаний з неактивною формою TGF-β, що зберігає його неактивність результуючи у відсутності біологічної активності останнього. Найпростіша модель синдрому Марфана виглядає приблизно так: зниження рівня фібриліну-1 спричинює підвищення рівня TGF-β через зворотній негативний зв'язок синтезу останнього з зв'язуванням з рецепторами. Хоча не є доведено взаємозв'язок TGF-β і специфічного патогенезу цього захворювання, проте доведено виникнення запальної реакції з вивільненням базофільних протеаз, що власне і спричиняє повільну деградацію еластичних волокон та інших компонентів екстрацелюлярного матриксу. Важливість TGF-β у формування цього патологічного механізму була доведена після відкриття синдрому Луїза-Діца, який спричинено змінами TGFβR2 гена 3 хромосоми, що кодує рецепторний протеїн до TGF-β.[17] Синдром Марфана часто плутається з синдромом Луїза-Діца, через практично однаковий клінічний перебіг обох патологій.[18]
Клінічні ознаки
Сьогодні відомо більше 30 різних ознак та симптомів, за допомогою яких можна діагностувати синдром Марфана. Найбільш виразні з них асоційовані з кістковою, серцево-судинною та зоровою системами, проте часто буває уражена вся сполучна тканина.
Кісткова система
Більшість видимих ознак пов'язані з кістковою системою. Багато хворих на синдром Марфана ростом вище середнього зріст, мають легку непропорційність, довгі, худі кінцівки, тонкі зап'ястя з довгими пальцями і ступнями (арахнодактилія). Окрім цього, хворі на синдром Марфана можуть мати сколіоз, грудний лордоз, деформовану груднину, високу гнучкість суглобів, високо розміщену піднебінну кістку з деформованим зубним рядом та порушенням прикусу, плоскостопість, сутулість і розтяжки на шкірі. Ці порушення можуть спричиняти біль в суглобах, кістках і м'язах. Деякі хворі на синдром Марфана мають порушення мовлення, яке спричинюють вади розвитку - високе піднебіння і малі щелепи. Часто у хворих виникає остеоартрит. Інші ознаки включають обмежений рух стегон, через атипове розміщення голівки стегнової кістки в кульшовому суглобі[19][20]
Зорова система
При синдромі Марфана порушення зорової системи різноманітні, проте в більшості зустрічається дислокація кришталика.[20] Це трапляється через слабкість війчастого пояска (циннової зв'язки) — сполучнотканинних тяжів, які взаєморозташовують кришталик в оці. Війчастий поясок містить у великій кількості фібрилін-1, що і створює підвищену пластичність пояска при порушеннях конформації білка. Нижні шари циннової зв'язки мають найбільш виражену дисфункцію, що власне і спричиняє рух кришталика вгору і назовні, у більшості хворих. При синдромі Марфана супутніми є близькозорість, проте трапляється і далекозорість, особливо у випадках дислокації кришталика всередину по відношенню до передньої камери ока. Сублюксація (часткова дислокація) кришталика визначається клінічно у 80 % пацієнтів за допомогою сліт-ламп біомікроскопа.
Інші ознаки та симптоми пов'язані з враженнями системи зору включають в себе збільшення кривини очного яблука, міопію, страбізм, ексотропію та езотропію.[20]
Серцево-судинна система
Життєво загрозливі ознаки і симптоми, пов'язані з порушеннями серцево-судинної системи, є: слабість, задишка, тахікардія (пришвидшене серцебиття), загруднинній біль, який інколи іррадіює в спину, плече або руку. При порушеннях периферичної гемодинаміки у хворих синдромом Марфана може бути виражена гіпотермія кінцівок. Порушення серцевих тонів, провідної системи серця, а також симптоматика серцевої ішемії повинні бути сигналом для лікаря до наступних обстежень серцево-судинної системи. Для хворих на синдром Марфана є типовим дегенерація медії аортального та мітрального клапанів, що спричиняє недостатність (регургітацію) останніх. Однак найважливішою ознакою для діагностики синдрому Марфана залишається аневризма (розширення) аорти. Відомі випадки безсимптомного перебігу аневризми аорти, яка після повної дегенерації медії розшаровувалась, що власне і спричиняє підвищену смертність хворих.
Через порушення сполучної тканини існує підвищений ризик дислокації мітрального протезу.[21] Через це надається перевага хірургічному ремоделюванню над протезуванням клапана. У випадку протезування аорти рекомендуються клапан-збережувальні операції (операція Девіда тощо).
Легені
Легенева симптоматика не є головною особливістю перебігу синдрому Марфана,[22] проте деколи може виникати спонтанний пневмоторакс.[23] Під час спонтанного одностороннього пневмотораксу, повітря потрапляє у плевральний простір між грудною кліткою і легенями. Через це виникає компресія і навіть колапс легень. Це супроводжується відчуттям хворого болю, задишки, ціанозу і може спричинити смерть. Інші легеневі прояви синдрому Марфана включають нічне апное[24] та ідіопатичні обструктивні легеневі захворювання.[25] Патологічні зміни у легенях включають кистозні зміни, емфізему, пневмонії, бронхіектаз, верхівковий фіброз і вроджені зміни, такі як гіпоплазія середньої частки.[22]
Нервова система
Дуральна ектазія — ослаблення сполучної тканини твердої оболони спинного мозку часто є причиною зниження якості життя хворого. Це ускладнення часто є недіагностоване, внаслідок відсутності явних ознак. Симптоматика наступна: біль у нижній частині спини, ногах, у животі, інші неврологічні симптоми у нижніх кінцівках та навіть головний біль. Ці симптоми більш виражені при горизонтальному положенні хворого. Дуральна ектазія в ранніх стадіях розвитку рідко діагностується за допомогою рентген-моніторингу. При магнітно-резонансному обстеженні проявляється розширеною твердою оболоною поперекових хребців.[26] Інші хребцеві проблеми, пов'язані з синдромом Марфана, включають дегенерацію хребцевих дисків, спінальні кисти і дисфункцію автономної нервової системи.
Генетика
Кожний з батьків має 50 % передачі генетичного дефекту кожному з дітей через автосомно-домінантну природу мутації. Більшість хворих на синдром Марфана мають родича з цим захворюванням. У близько 15-30 % усіх хворих дана мутація виникла вперше;[16] такі спонтанні мутації виникають в 1 з 20.000. Синдром Марфана є також прикладом негативної домінантної мутації і гаплонедостатності.[27][28] Ця мутація має варіабельну експресивність, неповна пенентрантність не була задокументована.
Діагностика
Діагностичні критерії синдрому Марфана були міжнародно затвердженими у 1996.[29] Діагноз синдрому Марфана базується на основі родинної історії і поєднання основних та другорядних індикаторів порушення, що рідко зустрічається в більшості популяції, наприклад: чотири ознаки порушень кісткової системи з однією або більше ознаками інших систем організму, таких як зорового аналізатора чи серцево-судинної, які одночасно можна діагностувати у одного хворого. Наступні стани можуть виникати внаслідок синдрому Марфана, проте зустрічаються у людей без цього синдрому.
Ревізія Гентської нозологічної класифікації
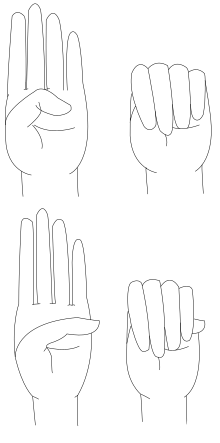
У 2010 нозологія Гента була переглянута і нові діагностичні критерії додалися до попередньої домовленості 1996. 7 нових критерій можуть бути використані для діагностики:[40][41]
У випадку відсутності родинної історії синдрому Марфана:
- Z-score кореня аорти ≥ 2 і вивих кришталика
- Z-score кореня аорти≥ 2 і мутація FBN1 гена
- Z-score кореня аорти ≥ 2 і системна оцінка* > 7 points
- Вивих кришталика і мутація FBN1 з діагностованою вадою аорти
У випадку наявності синдрому Марфана в родичів:
- Вивих кришталика
- Системна оцінка* ≥ 7
- Z-score кореня аорти≥ 2
- Системна оцінка:
- Зап'ясткова і долонна ознака = 3 (у випадку наявності лише однієї = 1)
- Pectus carinatum формація = 2 (pectus excavatum або асиметрія грудної клітки = 1)
- Плоскостопість = 2
- Дуральна ектазія = 2
- Protrusio acetabuli = 2
- Пневмоторакс= 2
- Зменшення співвідношення верхньої і нижньої частини тіла і збільшення співвідношення довжини рук до висоти і тяжкий сколіоз = 1
- Сколіоз або кіфоз = 1
- Знижена рухливість ліктя = 1
- Лицеві ознаки (3/5) = 1 (доліхосефалія, енофтальмоз, звисаючі повіки, гіпоплазія молярів, ретрогнатія)
- Розтяжки шкіри (стрії) = 1
- Міопія > 3 diopters = 1
- Пролапс мітрального задньої стулки мітрального клапана 1⁄4 1
Долонна ознака (ознака Стайнберга) визначається, якщо у пацієнта гіпермобільний ліктьово-зап'ястний і променево-зап'ястний суглоби.
Зап'ясткова ознака (ознака Волкера) позитивна, якщо пацієнт може обхопити однією рукою другу, торкаючись великим пальцем мізинця.
Диференційна діагностика
Багато різних порушень можуть продемонструвати симптоматику синдрому Марфана.[42] Генетичні дослідження і оцінка різних ознак та симптомів дозволяють їх відрізнити. Наступні захворювання схожі за перебігом на синдром Марфана:
- Синдром Ашара
- Вроджена арахнодактилія або синдром Білса
- Синдром Ехлера-Данлоса
- Гомоцистинурія
- Синдром Луїса-Дієца
- MASS фенотип
- Множинна ендокринна неоплазія, тип 2B
- Синдром Шпрінтцена-Голдберга[43]
- Синдром Стіклера
Лікування
Немає причинного лікування синдрому Марфана, проте впродовж останніх тривалість життя хворих значно покращилось і зараз схоже до життя звичайної людини.[44] Синдром лікується симптоматично, по відношенню до ушкоджень різних органів. У дітей медикаментозно запобігається дилатація (розширення) аорти.
Регулярний огляд у кардіолога є необхідним для спостереження серцевих клапанів і аорти. Головною ціллю у лікуванні є сповільнення прогресії дилатації аорти і ушкодження серцевих клапанів, за допомогою профілактики порушень провідної системи серця (аритмії, тахікардія) і зменшення системного кров'яного тиску.
Медикаментозна терапія
Медикаментозна терапія часто включає бета-блокатори, такі як пропранолол, або у разі нетолерантності хворого до нього, блокаторів кальцієвих каналів та ACE інгібіторів .[7][8]
Через те, що антагоністи ангіотензин-2 рецепторів також знижують вміст TGF-β. ці препарати досліджувались на пацієнтах з тяжким перебігом синдрому Марфана і спричинило сповільнення розширення аорти.[45] Проте, нещодавні дослідження показали схожі результати після використання ARB, лозартану і сучасної бета-блокаторної терапії, наприклад препарату атенолол.[46]
Фізична активність
Американська Асоціація Серця (The American Heart Association) має наступні рекомендації для хворих синдромом Марфана з відсутньою або малою дилятацією аорти:
- Скоріше дозволені заняття: боулінг, гольф, снорклінг, спортивне ходіння, тредміл.
- Середній ризик несуть наступні: баскетбол, рокетбол, сквош, біг, катання на лижах, футбол, теніс, бейсбол, катання на мотоциклі, кінний спорт.
- Високий ризик несуть наступні: бодібілдинг, вайтліфтинг, хокей, скелелазіння, віндсерфінг, серфінг, скубадайвінг.
Хірургічні втручання
Якщо дилятація аорти прогресує, викликаючи розриву або травми кореня аорти, а також призводить до тяжкої недостатності аортального або інших клапанів, тоді хірургічне втручання стає необхідним. Протезування аорти це складна операція, проте виконана планово має значно більше шансів на успіх. Саме втручання залежить від тяжкості стану пацієнта і зараз існують операції зі збереженням клапанного апарату.[47] Існує тенденція і до збільшення кількості інших судинних операцій у хворих на синдром Марфана, через збільшення тривалості життя останніх, наприклад: протезування низхідної частини грудної аорти а також інших судин.
Кісткові і зорові прояви синдрому Марфана також можуть бути складними, проте ніколи життєво небезпечними. Ці симптоми зазвичай лікуються типово, наприклад різними знеболювальними або м'язовими релаксантами. Через те, що синдром Марфана може спричинити асимптоматичні спинні деформації, різного роду хребтові операції мусять виконуватись з особливою обережністю, незважаючи на складність самої операції.
Лікування спонтанного пневмотораксу залежить від кількості повітря у плевральному просторі і перебігу ускладнення у конкретного хворого. Невеликий пневмоторакс може самовирішитись без активного втручання протягом 1-2 тижнів. Рецидивуючі пневмоторакси можуть потребувати хірургічного втручання. Тяжкі пневмоторакси вимагають грудної дренажної системи, яку використовують протягом декількох днів. Великі пневмоторакси є ургентними станами, які вимагають ургентної декомпресії.
Вагітність
Упродовж вагітності, навіть за відсутності серцево-судинних аномалій, жінки з синдромом Марфана мають високий ризик розшарування аорти, яке часто є фатальним за відсутності негайного хірургічного втручання. Жінки з синдромом Марфана повинні проходити ехокардіографічне обстеження кожні 6-10 тижнів упродовж вагітності, для оцінки діаметра кореня аорти. Під час періоду пологів більшості виконується кесарів розтин.[48]
Синдром Марфана експресується домінантно. Це означає, що дитина одного з батьків, який переносить мутований ген, має 50 % шанс народитись хворою. У 1996 році був виконаний перший передімплантаційний генетичний тест.[49] Це означає, що на ранньому етапі вагітності було проведене генетичне тестування, для визначення і переривання вагітності у випадку наявності в ембріона мутованого гена, відповідального за синтез фібриліна-1.
Прогноз
Завдяки сучасній серцевосудинній хірургії і терапії лозартаном і метопрололом, прогноз у хворих на синдром Марфана позитивний. Проте, раніше тривалість життя у вищезгаданих була знижена в середньому на третину. Більшість помирали у підлітковому віці через серцево-судинні проблеми. Сьогодні адекватне профілактичне лікування і рання дозволяє уникнути більшості симптомів. Отже існує тенденція до подовження життя хворих на синдром Марфана.[50] Відомо що жінки з Марфаном в середньому живуть довше.
Суспільство і культура
Популяризатори синдрому Марфана включають:
- Фло Гімен (Flo Hyman), олімпійська срібна медалістка з жіночого волейболу (1984), яка раптово померла під час матчу від розшарування аорти.[51]
- Джонатан Ларсон (Jonathan Larson), автор і композитор Рент (Rent), який помер від розшарування аорти, день перед музичним виступом.[52][53]
- Вінсент Шіавеллі (Vincent Schiavelli), актор і представницьке лице Фундації Марфана (The Marfan Foundation), пізніше перейменована в Національну Фундацію Марфана, який хворів на синдром Марфана, але помер від невідомої причини.
- Музикант Бредфорд Кокс (Bradford Cox) з інді-рок гурту Діргантер (Deerhunter).[54]
- Офшор Еван Робертсон (Ewan Robertson), музикант і графічний редактор Біуг Дада Рекордінгс (Big Dada Recordings), який раптово помер під час серцевої операції, пов'язаної з синдромом Марфана.[55]
- Ісаах Овстін (Isaiah Austin), гравець баскетбольної команди, діагностований з синдромом Марфана, через що мусів покинути кар'єру NBA(Ен-Бі-Ей).[56][57]
- Жавієр Ботет (Javier Botet), Іспанський актор, який грав ролі надприродних створінь у фільмах жахів, таких як: REC, Mama[58] і The Strain телевізійні серії.[59]
Дослідники вважають, що Ахенатен (Akhenaten), фараон Єгипту 18 покоління міг хворіти синдромом Марфана.[60][61]
Авраам Лінкольн (Abraham Lincoln) вважався протягом певного часу хворим на синдром Марфана,[62] проте ця точка зору була спростована сучасними генетиками[63][64]. Отримання точного результату є неможливим, через відмову отримання зразка ДНК.[65]
Примітки
- http://www.marfan.org/about/signs
- https://my.clevelandclinic.org/health/diseases/17209-marfan-syndrome/symptoms
- https://www.nhlbi.nih.gov/health-topics/marfan-syndrome
- http://omim.org/entry/154700
- What Is Marfan Syndrome?. NHLBI, NIH. 1 жовтня 2010. Процитовано 16 травня 2016.
- What Are the Signs and Symptoms of Marfan Syndrome?. NHLBI, NIH. 1 жовтня 2010. Процитовано 16 травня 2016.
- Marfan Syndrome. National Organization for Rare Disorders. 2014. Процитовано 16 травня 2016.
- How Is Marfan Syndrome Treated?. NHLBI, NIH. 1 жовтня 2010. Процитовано 16 травня 2016.
- Medical management of Marfan syndrome. Circulation 117 (21). 2008. с. 2802–13. PMID 18506019. doi:10.1161/CIRCULATIONAHA.107.693523.
- Marfan, Antoine (1896). Un cas de déformation congénitale des quartre membres, plus prononcée aux extrémitiés, caractérisée par l'allongement des os avec un certain degré d'amincissement [A case of congenital deformation of the four limbs, more pronounced at the extremities, characterized by elongation of the bones with some degree of thinning]. Bulletins et memoires de la Société medicale des hôspitaux de Paris (фр.) 13 (3rd series). с. 220–226.
- Antoine Bernard-Jean Marfan. Whonamedit?. Процитовано 16 травня 2016.
- Johns Hopkins Comprehensive Marfan Center. Архів оригіналу за 15 жовтня 2008. Процитовано 27 вересня 2016.
- Brown P (July 27, 1991).
- Determinants of quality of life in Marfan syndrome. Psychosomatics 49 (3). 2008. с. 243–8. PMID 18448780. doi:10.1176/appi.psy.49.3.243. Архів оригіналу за 13 липня 2012. Процитовано 27 вересня 2016.
- McKusick V (1991). The defect in Marfan syndrome. Nature 352 (6333). с. 279–81. Bibcode:1991Natur.352..279M. PMID 1852198. doi:10.1038/352279a0.
- Cotran; Kumar, Collins (1998). Robbins Pathologic Basis of Disease. Philadelphia: W.B Saunders Company. ISBN 0-7216-7335-X.
- Entrez Gene (2007). TGFBR2 transforming growth factor, beta receptor II (Entrez gene entry). NCBI. Процитовано 11 січня 2007.
- Related Disorders: Loeys-Dietz. National Marfan Foundation. Архів оригіналу за 25 вересня 2006. Процитовано 11 січня 2007.
- Van de Velde, S; Fillman, R; Yandow, S (2006). Protrusio acetabuli in Marfan syndrome. History, diagnosis, and treatment.. The Journal of bone and joint surgery. American volume 88 (3). с. 639–46. PMID 16510833. doi:10.2106/JBJS.E.00567.
- OMIM Entry - # 154700 - MARFAN SYNDROME; MFS. omim.org. Процитовано 8 серпня 2016.
- Zipes, Libby Bonow Braunwald (2005). Braunwald's Heart Disease ~ A Textbook of Cardiovascular Medicine, Seventh Edition. United States of America: Elseview Saunders. с. 1894. ISBN 0-7216-0509-5.
- Dyhdalo, K; Farver, C (2011). Pulmonary histologic changes in Marfan syndrome: a case series and literature review.. American journal of clinical pathology 136 (6). с. 857–63. PMID 22095370. doi:10.1309/AJCP79SNDHGKQFIN.
- Siepe, M; Löffelbein, F (2009). [The Marfan syndrome and related connective tissue disorders].. Medizinische Monatsschrift für Pharmazeuten 32 (6). с. 213–9. PMID 19554831.
- Kohler, M.; Blair, E.; Risby, P.; Nickol, A. H.; Wordsworth, P.; Forfar, C.; Stradling, J. R. (1 лютого 2009). The prevalence of obstructive sleep apnoea and its association with aortic dilatation in Marfan's syndrome. Thorax 64 (2). с. 162–166. ISSN 1468-3296. PMID 18852161. doi:10.1136/thx.2008.102756.
- Corsico, A. G.; Grosso, A.; Tripon, B.; Albicini, F.; Gini, E.; Mazzetta, A.; Di Vincenzo, E. M.; Agnesi, M. E. та ін. (1 червня 2014). Pulmonary involvement in patients with Marfan Syndrome. Panminerva Medica 56 (2). с. 177–182. ISSN 1827-1898. PMID 24994580.
- Marfan Syndrome. Mayo Clinic. Процитовано 12 січня 2007.
- Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. The Journal of Clinical Investigation 114 (2). 2004. с. 172–81. PMC 449744. PMID 15254584. doi:10.1172/JCI20641.
- Marfan's syndrome. Lancet 366 (9501). 2005. с. 1965–76. PMC 1513064. PMID 16325700. doi:10.1016/S0140-6736(05)67789-6.
- Revised diagnostic criteria for the Marfan syndrome. Am. J. Med. Genet. 62 (4). 1996. с. 417–26. PMID 8723076. doi:10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R.
- Marfan syndrome. Long-term survival and complications after aortic aneurysm repair. Circulation 91 (3). 1995. с. 728–33. PMID 7828300. doi:10.1161/01.CIR.91.3.728.
- Marfan Syndrome: Signs and Symptoms. www.ucsfhealth.org. Процитовано 28 серпня 2009.
- What is Marfan Syndrome?. Marfan Trust. Архів оригіналу за 10 червня 2015. Процитовано 1 червня 2015.
- Marfan Syndrome: The Similarities to Copper Deficiency. www.ctds.info. Процитовано 29 серпня 2009.
- MedlinePlus Encyclopedia Marfan syndrome
- Marfan syndrome. Genetics Home Reference. U.S. National Institute of Health. Процитовано 28 серпня 2009.
- The bone mineral status of patients with Marfan syndrome. Journal of Bone and Mineral Research 10 (10). 1995. с. 1550–5. PMID 8686512. doi:10.1002/jbmr.5650101017.
- Northwestern Memorial Center for Heart Valve Disease.
- About Marfan Syndrome: Features. National Marfan Foundation. Архів оригіналу за 20 серпня 2009. Процитовано 28 серпня 2009.
- Living with Marfan Syndrome: Dental issues. National Marfan Foundation. Архів оригіналу за 6 вересня 2009. Процитовано 28 серпня 2009.
- 2010 Revised Ghent Nosology. National Marfan Foundation. Архів оригіналу за 14 січня 2011. Процитовано 31 січня 2011.
- Loeys, BL; Dietz, HC; Braverman, AC; Callewaert, BL; De Backer, J; Devereux, RB; Hilhorst‑Hofstee, Y; Jondeau, G та ін. (2010). The revised Ghent nosology for the Marfan syndrome (PDF). Journal of Medical Genetics 47 (7) (London: BMJ Group). с. 476–485. ISSN 0022-2593. OCLC 857424767. PMID 20591885. doi:10.1136/jmg.2009.072785. Архів оригіналу за 10 січня 2016. Процитовано 27 вересня 2016.
- Emery and RImoin's Principles and Practice of Medical Genetics. 5th ed. Philadelphia, Pennsylvania: Churchill Livingstone Elsevier. 2007.
- Greally & GeneReviews, 2010
- Questions and Answers about Marfan Syndrome. Niams.nih.gov. Процитовано 23 червня 2014.
- Pyeritz RE (2008). A small molecule for a large disease. NEJM 358 (26). с. 2829–31. PMID 18579819. doi:10.1056/NEJMe0804008.
- R.V. Lacro et al.: Atenolol versus Losartan in Children and Young Adults with Marfan's Syndrome.
- Heart Surgery for Marfan Syndrome. Mayo Clinic. Архів оригіналу за 18 грудня 2006. Процитовано 12 січня 2007.
- Chen H (2007). Marfan Syndrome. eMedicine. Процитовано 25 червня 2007.
- Preimplantation genetic testing for Marfan syndrome. Mol. Hum. Reprod. 2 (9). 1996. с. 713–5. PMID 9239687. doi:10.1093/molehr/2.9.713.
- Keane, Martin G.; Pyeritz, Reed E. (2008). Medical Management of Marfan Syndrome. Circulation 117 (21). с. 2802–2813. ISSN 1524-4539. PMID 18506019. doi:10.1161/CIRCULATIONAHA.107.693523.
- Flo Hyman. Volleyball Hall of Fame. Архів оригіналу за 30 січня 2008. Процитовано 6 січня 2009.
- Lawrence Van Gelder (13 грудня 1996). On the Eve of a New Life, an Untimely Death. The New York Times. Процитовано 17 липня 2008.
- Kirk, Fiona J. (26 липня 2011). Syndrome survival: New drugs offer promise for often-fatal Marfan tissue disorder. The Daily. Процитовано 24 листопада 2011.
- Deerhunter interview. Pitchfork.com. 11 червня 2007. Процитовано 1 жовтня 2014. «(Interviewer): i think a lot of people still don't know [that you have Marfan Syndrome]... (Bradford Cox): People think I'm a junkie.»
- Big Dada to release final, posthumous album by Offshore. FACT Magazine. 14 липня 2015. Процитовано 13 серпня 2016.
- NBA makes Austin's dreams come true with gesture at draft. Houston Chronicle. 26 червня 2014.
- Isaiah Austin has Marfan syndrome. ESPN.com. 22 червня 2014. Процитовано 22 червня 2014.
- Chang, Justin (15 січня 2013). Mama. Variety.
- The Creature and Makeup Effects of The Strain - Tested.com. Tested. Процитовано 9 лютого 2016.
- The Mystery of Akhenaten: Genetics or Aesthetics?.
- Akhenaten's illness.
- Marion R (1994). Mr. Lincoln and Dr. Marfan's syndrome. Was George Washington Really the Father of Our Country?. Reading, MA: Addison-Wesley.
- Sotos, John G. The Physical Lincoln: Other Medical Theories. www.physical-lincoln.com. Mt. Vernon Book Systems. Процитовано 13 серпня 2016. «Medical Condition: Marfan syndrome; Did Lincoln Have It? No»
- Sotos JG (2008). The Physical Lincoln Sourcebook. Mt. Vernon, VA: Mt. Vernon Book Systems. с. 29. ISBN 978-0-9818193-3-4.
- Ready T (1999). Access to presidential DNA denied. Nature Medicine 5 (8). с. 859. PMID 11645164. doi:10.1038/11287.