Ішемічний інсульт
Ішемічний інсульт (інфаркт мозку) — гостре порушення мозкового кровообігу внаслідок дефіциту надходження артеріальної крові до головного мозку, що призводить до його гіпоксії та розвитку ділянок некрозу. Виникає переважно внаслідок оклюзій кровоносних судин.
| Ішемічний інсульт (інфаркт мозку) | |
|---|---|
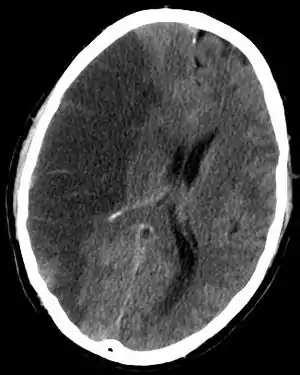 Комп'ютерна томограма головного мозку, яка демонструє інфаркт правої півкулі головного мозку (на зображенні розташований зліва) Комп'ютерна томограма головного мозку, яка демонструє інфаркт правої півкулі головного мозку (на зображенні розташований зліва) | |
| Спеціальність | неврологія |
| Препарати | dantrolened[1], Alteplased[2], Аторвастатин[2], Ривароксабан[2], Цилостазол[2] і клопідогрель[2] |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | 8B11.5 |
| МКХ-10 | I63 |
| OMIM | 601367 |
| eMedicine | neuro/ |
| MeSH | D002544 |
| SNOMED CT | 432504007 |
| | |
Причини
Під час аналізу, заснованого на сукупності дослідження, проведеного в групі хворих в Оксфорді (n = 209), була створена оцінна шкала ABCD для визначення високого ризику інсульту (Rothwell, 2005; клас B). При комплексному аналізі виділено 5 основних незалежних факторів високого ризику розвитку ІІ:
- вік старше 60 років;
- цукровий діабет;
- ТІА (транзиторна ішемічна атака), що триває понад 10 хвилин;
- ТІА зі слабкістю (один із симптомів);
- ТІА з порушенням мови.
Патогенез
Патофізіологічні механізми загибелі нервових клітин
Під час ішемічного інсульту клітина може загинути двома основними шляхами — некрозом та апоптозом[3][4][5][6]. Перший шлях домінує в гостру фазу ішемічного інсульту (перші декілька хвилин) в ділянці з найбільш критичним кровопостачанням. Ця ділянка в подальшому стане ядром інфаркту.[4][6][7][8][9] Довкола ядра інфаркту розташована пенумбра — ділянка, у якій ішемія не настільки критична. Нейрони тут функціонально неактивні, проте зберігають свою клітинну цілісність[7][10]. У пенумбрі клітини можуть загинути двома способами (як некрозом, так і апоптозом), адже патологічні реакції, які йдуть у ішемізованій клітині, є неспецифічними для якогось певного шляху[5][11][12], проте панівним шляхом є саме апоптоз[6][9][12][13]. Апоптоз також є домінантним шляхом у випадку віддаленої загибелі клітин, як може виникнути у ділянці, яка короткочасно була піддана ішемії[5]. Певна частина клітин також гинуть шляхом некроптозу[3][5] та автофагії[5][14].
Стадії патогенезу
Розрізняють три фази у патогенезі ішемічного інсульту:
- гостру (до декількох хвилин) — клітини набрякають, їхні мітохондрії руйнуються, клітини гинуть та формують ядро інфаркту;
- підгостру (до 6 годин) — явище періінфарктної деполяризації, збільшення розмірів ядра інфаркту за рахунок пенумбри;
- віддалену (від декількох днів до декількох тижнів) — вазогенний набряк, запалення, віддалений апоптоз.
Ішемічний каскад
Ішемічний каскад — це сукупність реакцій, які виникають за умов ішемії та ведуть до пошкодження клітин мозку. Ці реакції можуть привести як до некрозу, так і до розвитку апоптозу. Вони йдуть як у ядрі інфаркту, так і у пенумбрі, проте різняться тривалістю перебігу.[5][11][15]
У нормі нейрони велику кількість енергії використовують для функціонування іонних насосів, найважливішим з яких є натрій-калієвий насос. Це забезпечує стабільну концентрацію іонів у клітині та поза нею, що потрібно для збудження нейронів (деполяризації та реполяризації). Проте ці насоси потребують енергії яка утворюється при розщепленні зв'язків АТФ. В умовах ішемії (порушення постачання кисню, глюкози та інших поживних речовин) синтез АТФ порушується, що в свою чергу веде до порушення в іонному гомеостазі — аноксичної деполяризації (зміни заряду мембрани в умовах нестачі кисню).[10][16][17] Калій, якого багато у клітині, виходить з неї, а натрій та хлор, яких багато поза клітиною, прямують у клітину. Надмір натрію провокує підвищення осмолярності цитоплазми клітини, що у свою чергу веде до входження у клітину великої кількості води — розвивається цитотоксичний набряк, або набухання клітини.[18][19] Проте цитотоксичний набряк сам не викликає набряк мозку, який можна візуалізувати на КТ чи МРТ, а лише після того, як до нього приєднаються іонний та вазогенний набряки. Перший виникає разом із набуханням клітин, якщо збережений хоча б якийсь кровотік (найхарактерніше для пенумбри) та за непошкоженого гемато-енцефалічного бар'єру (ГЕБ). Його суть полягає в тому, що переміщення натрію та хлору в клітину спричиняє дефіцит цих іонів у позаклітинному середовищі, який нівелюється надходженням іонів з кровоносних судин. Ці іони «тягнуть» за собою воду.[16][20] Через 4–6 годин пошкоджуються елементи ГЕБ, велика кількість рідини прямує в позаклітинний простір і розвивається таке пізнє ускладнення, як вазогенний набряк. Свого піку він сягає на 2-4 добу та суттєво збільшує об'єм мозку. Вазогенний набряк у свою чергу погіршує перфузію та може провокувати дислокацію мозку.[16][19]
Деполяризація спричиняє викид нейронами із своїх пресинаптичних закінчень великої кількості глутамату — збудливого нейромедіатора, який дуже поширений в ЦНС. Його стає дуже багато (нейроглія, яка його поглинає після виділення, також пошкоджена), а у великих кількостях він є токсичним. У випадку глутамату така токсичність називається ексайтотоксичністю: медіатор приєднюється до NMDA- та AMPA-рецепторів та провокує вхід великої кількості кальцію, який у свою чергу активує велику кількість ферментів (фосфоліпаз, ендонуклеаз, протеїнкіназ та протеаз).[21][22][23] Загалом ексайтотоксичність запускає ланцюг реакцій, які наведені нижче.
- Активовані ферменти пошкоджують клітинні структури, а також запускають пероксидне окислення з утворенням вільних радикалів, які ще більше пошкоджують клітину[18][21].
- Вільні радикали та надмір кальцію пошкоджують мітохондрії — «енергетичний» центр клітини. Залежно від інтенсивності ішемії (ядро інфаркту чи пенумбра), мітохондрії можуть ушкоджуватися повністю (веде до некрозу), або частково, коли через збільшену проникність з них виділяються проапоптичні фактори (наприклад, апоптоз-індукуючий фактор)[5][19][24].
- Ексайтотоксичність також провокує стрес еноплазматичного ретикулума, який може проявлятися пригніченням синтезу білка чи синтезом стресових білків та білків неправильної будови. Наявність неправильних білків також провокує апоптоз.[19][25]
- Разом з кальцієм у клітину заходить велика кількість цинку, який у свою чергу потенціює утворення вільних радикалів та ушкодження мітохондрій, а також, незалежно від мітохондріальної дисфункції, запускає апоптоз[21][22].
Вище перелічені механізми належать до внутрішніх механізмів активації апоптозу. Окрім внутрішніх механізмів, апоптоз у зоні ішемії також провокується зовнішніми механізмами — стимуляцією рецепторів Fas, TNFαR, а в зоні ішемії ще й TLR2, TLR4 та NOTCH-1.[19][26]
Кальцій потрапляє в клітину й іншим, незалежним від глутамату, шляхом — активацією протонактивованих іонних каналів (канали, які активуються при більш кислому середовищі). Кислим середовище в клітині стає через те, що при дефіциті кисню, окислення глюкози закінчується анаеробним шляхом, при якому окрім двох молекул АТФ утворюється піруват. Глутамат також стимулює виділення кальцію, який у великих кількостях міститься в ендоплазматичному ретикулумі. Таким чином, рівень кальцію у клітині зростає завдяки декільком механізмам.[11][27][22]
На перебіг інсульту впливає ще один патофізіологічний механізм — запалення. Свого піку воно сягає на сьому добу. У місці ішемії відбувається підвищена експресія генів, які кодують NF-κB, TNF-α, IL-1β, IL-6, білки теплового шоку. Спочатку активується мікроглія, яка виконує роль імунної системи в головному мозку, а згодом в речовину мозку потрапляються чужорідні клітини — нейтрофіли, лімфоцити, моноцити та макрофаги. Останні є найчисленнішими клітинами на сьомий день інсульту. У мозок вони потрапляють завдяки підвищеній експресії генів, що кодують молекули міжклітинної адгезії (виробляються ендотелієм).[5][18][19][28] Запалення має негативний вплив: імунні клітини виділяють вільні радикали, які ще сильніше пошкожують і так пошкоджену тканину.[5][28]
Клінічні ознаки
При тромбозі розвиток захворювання поступовий, часто під час сну. Відмічається період передвісників у вигляді запаморочення, короткочасної слабкості та оніміння в кінцівках. Вогнищеві симптоми розвиваються зазвичай при збереженій свідомості. При інсульті в басейні середньої мозкової артерії спостерігається déviation conjuguée.
Емболія виникає при ендокардитах, миготливій аритмії. Вогнищеві симптоми з'являються швидко, часто з виникненням короткочасної втрати свідомості, іноді фокальні судомні напади.
Діагностика
Пацієнт повинен бути невідкладно обстежений на місці (клінічна картина ТІА або іншого захворювання) або при вступі в стаціонар. У стаціонарі обов'язкові такі діагностичні заходи:
- якщо симптоматика припускає наявність ішемії в зоні сонної артерії, проводиться ультразвукове дослідження (УЗД), КТ-ангіографія або магнітно-резонансна ангіографія (МРА);
- контроль серцевого ритму;
- при підозрі на кардіоемболію — ехокардіографія (ЕхоКГ).
Лабораторні аналізи:
- загальний аналіз крові;
- визначення рівня глюкози;
- визначення електролітів (натрій, калій, хлор, CO2);
- визначення швидкості осідання еритроцитів (ШОЕ).
Електрокардіографія (ЕКГ).
Візуалізація головного мозку і судин (Douglas, 2003; клас D):
- магнітно-резонансна томографія (МРТ)/МРА
- КТ/КТ-ангіографія
- КТ, УЗД сонної артерії при підозрі на її оклюзію.
Лікування
Специфічна терапія гострого ішемічного інсульту включає:
- системний тромболізис шляхом в/в введення рекомбінованого тканинного активатора плазміногену;
- антитромбоцитарну терапію ацетилсаліциловою кислотою;
- лікування набряку головного мозку та підвищеного ВЧТ з допомогою сорбілакту, манітолу або гіпертонічного розчину натрію хлориду (10 %);
- нейроендоваскулярні хірургічні втручання;[29]
- едаравон шляхом внутрішньовенної інфузії двічі на добу протягом двох тижнів[30][31]
Протипоказане введення нефракціонованого гепарину, гепарину низької молекулярної маси і гепариноїдів.[29]
Не рекомендується для лікування пацієнтів в гострому періоді інсульту застосування лікарських засобів, використання яких може мати негативні наслідки або негативно вплинути на клінічний перебіг гострого періоду ішемічного інсульту, а саме:
- Розчинів глюкози.
- Діуретиків (осмотичних у вигляді розчинів та фуросеміду).
- Колоїдних розчинів для гемодилюції.
- Вазоактивних препаратів (препарати барвінку, пентоксифілін).
- Препаратів блокаторів кальцієвих каналів короткої дії для корекції системного АТ (ніфедипін) та корекції внутрішньомозкової гемодинаміки в гострому періоді ІІ (німодипін).[29]
Не підтверджена в гострому періоді ішемічного інсульту клінічна ефективність призначення спазмолітиків, антиоксидантів, препаратів, які впливають на метаболізм (мілдронат), препаратів бурштинової кислоти, токоферолу, ноотропних засобів, глюкокортикоїдів, нейропротекторів.[29]
Прогноз
Визначається локалізацією і об'ємом інфаркту, виразністю набряку мозку, а також наявністю супутніх захворювань і / або розвитком ускладнень протягом інсульту (пневмонія, пролежні, уросепсис і ін.). У перші 30 днів помирає близько 15-25% хворих. Смертність вище при атеротромботичному та кардіоемболічному інсультах, і становить лише 2% при лакунарному.
Тяжкість і прогресування інсульту часто оцінюють, використовуючи стандартизовані вимірювачі, наприклад, шкалу інсульту Національного Інституту Здоров'я (NIH) [60].
Причина смерті в половині випадків - набряк мозку і викликана ним дислокація структур мозку, в інших випадках - супутні тяжкі ускладнення, як от пневмонія, серцеві захворювання, емболія легеневої артерії, ниркова недостатність або септицемія. Значна частина (40%) летальних результатів виникає в перші 2 доби захворювання і пов'язана з великими розмірами зони інфаркту та набряком мозку.
Ішемічний інсульт, за даними досліджень Британського інституту медицини, в 3 з 10 випадків у сукупності з атеросклерозом (особливо характерно для людей від 75 років), може паралізувати роботу легенів через недостатній кровообіг в них, що, через 30-90 днів після перенесеного інсульту, призведе до обструкції легенів, кашлю, гарячки, та в кінцевому рахунку викличе пневмонію або розмноження бактерій. Внаслідок цього може пройти відмирання і / або роз'їдання легеневих тканин, і легені не зможуть більше виконувати свої функції, що призведе до смерті.
У перший місяць після ішемічного інсульту близько 60-70% хворих, які вижили, мають значні інвалідизуючі неврологічні розлади. Через 6 місяців після інсульту інвалідизуючі неврологічні розлади залишаються у 40% хворих, які вижили, до кінця року - у 30%. Чим більш значний неврологічний дефіцит спостерігається до кінця першого місяця після інсульту, тим менш імовірне повне відновлення.
Повторний ішемічний інсульт виникає приблизно у 30% хворих в період 5 років після першого інсульту
Виноски
- NDF-RT
- Drug Indications Extracted from FAERS — doi:10.5281/ZENODO.1435999
- Grotta, 2015, с. 62.
- Brainin, 2014, с. 7-8.
- Matthias Endres, Ulrich Dirnagl, Michael A. Moskowitz (2008). The ischemic cascade and mediators of ischemic injury. Handbook of Clinical Neurology (англ.) 92: 31–41.
- Andrew Bivard; Neil Spratt; Christopher R Levi; Mark W Parsons (2011). Time to Dispense with the Clock and Move to Tissue-based Decision Making?. Expert Review of Cardiovascular Therapy (англ.) 9 (4): 451–461.
- Caplan, 2009, с. 50.
- Brad R.S. Broughton; David C. Reutens; Christopher G. Sobey (2009). Apoptotic Mechanisms After Cerebral Ischemia. Stroke (англ.) (40): 331–339. doi:10.1161/STROKEAHA.108.531632.
- Natan Bornstein. Stroke: Practical Guide for Clinicians. — Karger, 2009. — 24-25 с. — ISBN 978-3805590990.
- Яворська В.О., Фломін Ю.В. Специфічне лікування ішемічного інсульту: нейропротекція. Новости медицины и фармации. Архів оригіналу за 10 листопада 2015. Процитовано 10 листопада 2015.
- Brainin, 2014, с. 18.
- Grotta, 2015, с. 68.
- Tiina Sairanen, Marja-Liisa Karjalainen-Lindsberg, Anders Paetau, Petra Ija¨s and Perttu J. Lindsberg (2006). Apoptosis dominant in the periinfarct area of human ischaemic stroke — a possible target of antiapoptotic treatments. Brain (англ.) 129: 189–199. doi:10.1093/brain/awh645.
- Grotta, 2015, с. 67.
- Alexander Kunz, Ulrich Dirnagl, Philipp Mergenthaler (2010). Acute pathophysiological processes after ischaemic and traumatic brain injury. Best Practice & Research Clinical Anaesthesiology (англ.) 24 (4): 495–509. doi:10.1016/j.bpa.2010.10.001.
- Alexander S. Thrane, Vinita Rangroo Thrane, Maiken Nedergaard (2014). Drowning stars: reassessing the role of astrocytes in brain edema. Trends in Neuroscience (англ.) 37 (11): 620–628. doi:10.1016/j.tins.2014.08.010.
- Brainin, 2014, с. 12-14.
- Prabal Deb, Suash Sharma, K.M. Hassan (2010). Pathophysiologic mechanisms of acute ischemic stroke: An overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology (англ.) 17 (3): 197–218. doi:10.1016/j.pathophys.2009.12.001.
- Brainin, 2014, с. 20-21.
- Kristopher T. Kahle, J. Marc Simard, Kevin J. Staley, Brian V. Nahed, Pamela S. Jones, Dandan Sun (2009). Molecular Mechanisms of Ischemic Cerebral Edema: Role of Electroneutral Ion Transport (англ.) 24 (4). с. 257–265. doi:10.1152/physiol.00015.2009.
- Brainin, 2014, с. 19.
- Grotta, 2015, с. 71-72.
- В.О. Малахов, В.О. Монастирський, В.С. Личко, Г.М. Завгородня, І.Р. Скрипченко, А.В. Гетманенко (2011). Патогенетичні ланки ішемічного інсульту. Новости медицины и фармации. Процитовано 12 листопада 2015.
- Н. Р. Сохор, С. І. Шкробот (2014). Мітохондріальна дисфункція у гострий період ішемічного інсульту. Український неврологічний журнал 3–4 (32-33): 22–27.
- Scott A. Oakes, Feroz R. Papa (2015). The Role of Endoplasmic Reticulum Stress in Human Pathology. Annual Review of Pathology: Mechanisms of Disease (англ.) 10: 173–194. doi:10.1146/annurev-pathol-012513-104649.
- Grotta, 2015, с. 63-66.
- Grotta, 2015, с. 68-70.
- Grotta, 2015, с. 75-76.
- Міністерство охорони здоров’я (2012). УНІФІКОВАНИЙ КЛІНІЧНИЙ ПРОТОКОЛ МЕДИЧНОЇ ДОПОМОГИ "Ішемічний інсульт". http://mtd.dec.gov.ua/index.php/uk/. Міністерство охорони здоров’я. Процитовано 12 лютого 2019.
- Сучасні погляди на лікування ішемічного інсульту. Медична газета "Здоров'я України". 1 грудня 2018. Процитовано 12 лютого 2019.
- Watanabe, Kazutoshi (2018). Насколько эффективен эдаравон при лечении острого ишемического инсульта и бокового амиотрофического склероза? (російською). Міжнародний неврологічний журнал 6 (100). Процитовано 12 лютого 2019.
Джерела
- Michael Brainin, Wolf-Dieter Heiss. Textbook of Stroke Medicine. — Cambridge : Cambridge University Press, 2014. — 421 с. — ISBN 978-1107047495. (англ.)
- Bo Norrving. Oxford Textbook of Stroke and Cerebrovascular Disease. — Oxford : Oxford University Press, 2014. — 304 с. — ISBN 978-0199641208. (англ.)
- Louis Caplan. Caplan's Stroke: A Clinical Approach. — 4th. — Philadelphia : Saunders, 2009. — 688 с. — ISBN 978-1416047216. (англ.)
- James C. Grotta, Gregory W Albers, Joseph P Broderick, Scott E Kasner, Eng H Lo, A David Mendelow, Ralph L Sacco, Lawrence. Stroke: Pathophysiology, Diagnosis, and Management. — 6th. — Elsevier, 2015. — 1504 с. — ISBN 978-0323295444. (англ.)
- Julien Bogousslavsky, Louis R. Caplan. Stroke Syndromes. — 2nd. — Cambridge : Cambridge University Press, 2001. — 770 с. — ISBN 978-0521771429. (англ.)
