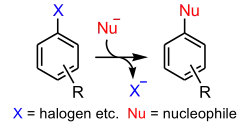
Нуклеофільне ароматичне заміщення
Нуклеофільне ароматичне заміщення (англ. nucleophilic aromatic substitution) — серія механізмів, що описують реакції нуклеофільного заміщення на ароматичному кільці. Формально цей тип реакцій можна віднести до реакцій нуклеофільного заміщення на ненасиченому атомі вуглецю, однак через особливі властивості ароматичних сполук йому зазвичай присвячується окремий розділ. Основні принципи нуклеофільного заміщення описані у відповідній статті:
Механістичні особливості
Через плоску структуру ароматичних кілець реакція ніколи не протікає за механізмом SN2: тилова нуклеофільна атака має відбуватись з центру кільця, що не є можливим.[1]

Водночас, механізм SN1 спостерігається лише у виключних випадках, оскільки феніл-катіон є енергетично невигідним через відсутність будь-якої стабілізації:[1]
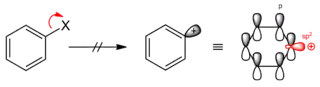
Механістичною альтернативою може бути механізм приєднання-елімінування, як і у випадку нуклеофільного ацильного заміщення, але існують також інші механізми; повну класифікацію представлено нижче:
- Приєднання-елімінування (скорочено SNAr);
- Елімінування-приєднання (також відоме як аріновий механізм);
- Механізм SN1 (через феніл-катіон) (напр. реакція Шимана);
- Вікарне нуклеофільне заміщення (VNS);
- Механізм ANRORC;
- Радикально-нуклеофільне ароматичне заміщення (SRN1, напр. реакція Зандмеєра).
Механізм приєднання-елімінування (SNAr)
Загальну схему механізму SNAr представлено нижче на прикладі заміщеного бензену:
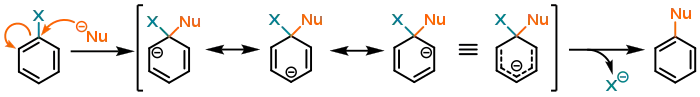
Під час атаки нуклеофіл взаємодіє з вільною π*-орбіталлю ароматичного кільця; завдяки цьому стає можливим нуклеофільне приєднання — але водночас порушується ароматичність. Утворений карбаніон є ізоелектронним із лінійним пентадієніл-аніоном.[1] Таким чином, нуклеофільне ароматичне заміщення є енергетично невигідним, якщо відповідний карбаніон не зазнає належної стабілізації в позиціях орто- і пара-, і такі реакції будуть проходити лише за дуже жорских умов. Найкраще негативний заряд стабілізують електроноакцепторні групи (-NO2 > -CN > -CHO > -CRO),[2] а електронодонорні групи, навпаки, дестабілізують його. Це є дзеркальною протилежністю до властивостей диригуючих груп в електрофільному ароматичному заміщенні:
| -NO2 | -CN | -CHO | -CRO | -NR2 | -OR | -OH | ||
|---|---|---|---|---|---|---|---|---|
| SNAr | Активуюча | ✓ | ✓ | ✓ | ✓ | |||
| Дезактивуюча | ✓ | ✓ | ✓ | |||||
| Позиція, куди диригує | орто-/пара- | орто-/пара- | орто-/пара- | орто-/пара- | мета- | мета- | мета- | |
| SEAr | Активуюча | ✓ | ✓ | ✓ | ||||
| Дезактивуюча | ✓ | ✓ | ✓ | ✓ | ||||
| Позиція, куди диригує | мета- | мета- | мета- | мета- | орто-/пара- | орто-/пара- | орто-/пара- |
У випадку влучно підібраного субстрату з електроноакцепторними групами (EWG) в орто- і/або пара-положенні до відхідної групи можливе утворення особливих реактивних інтермедіатів — комплексів Мейзенгеймера — які були підтверджені спектроскопічним шляхом або навіть ізольовані.[3][4][5] Вони виникають, коли EWG може приймати на себе негативний заряд, утворюючи подвійний зв'язок із атомом вуглецю, з яким вона пов'язана:
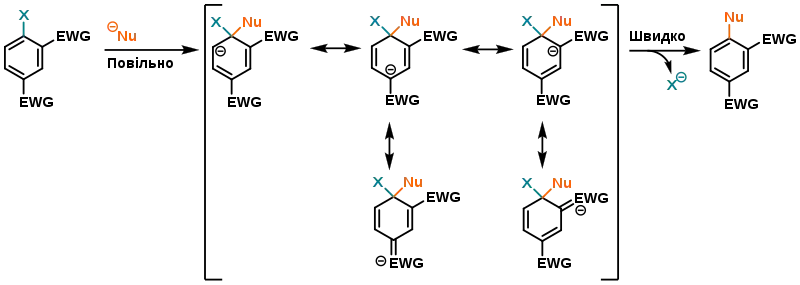
Тим не менш, комплекси Мейзенгеймера мають вищу потенціальну енергію, аніж вихідний субстрат, тому процес приєднання нуклеофілу проходить повільно — цей крок і є швидкістьвизначальним.[1] На відміну від реакцій аліфатичного нуклеофільного заміщення, тут відхідна група відіграє іншу роль, що підтверджується спадом швидкості реакцій SNAr при Х = F >> Cl > Br > I,[6][7] що є прямою протилежністю очікуваній реактивності у випадку аліфатичного заміщення. Це зумовлено тим, що більш електронегативі відхідні групи сильніше поляризують зв'язок C–X, що робить стадію приєднання більш сприятливою.[1]
Однак, при тому, що серед галогенів така закономірність зберігається завжди, в більш широкому сенсі швидкість реакції ще залежить від інших замісників, а також від нуклеофілу. Так, при нуклеофільному заміщенні на 1-Х-2,4-динітробензені швидкість реакції спадає в ряду Х = F ~ NO2 > OC6H4NO2(p) > Cl > OC6H5 > SC6H4NO2(p), якщо в якості нуклеофілу виступає метоксид MeO-, і Х = NO2 >> F > Cl > OC6H4NO2 ~ SC6H4NO2 > OC6H5, якщо в якості нуклеофілу виступає тіофенолят PhS-.[8]
Механізм елімінування-приєднання
Механізм елімінування-прирєднання (англ. elimination-addition mechanism),[1] також відомий як бензіновий механізм (англ. benzyne mechanism),[9] має місце у випадках реакцій галогеноаренів з нуклеофілом в присутності дуже сильної основи. Цей механізм має дві основні відмінності від SNAr:
- Така реакція призводить не тільки до іпсо-заміщення, а також і до т. зв. сіне-заміщення,[10] що пояснюється утворенням арінового інтермедіату — 1,2-дегідробензену, або бензіну (не плутати з бензином):

- Швидкість реакції спадає в послідовності Х = Br > I > Cl > F (при проведенні реакції із амідом калію в рідкому амоніаку). Це свідчить проти механізму SNAr.[11] Така дивна послідовність пояснюється зміною в природі лімітуючого кроку реакції: у випадку броміду і йодиду лімітуючим кроком є відщеплення відхідної групи (як у випадку SNAr, швидкість реакції має спадати в порядку F > Cl > Br > I), а у випадку хлориду і фториду — відщеплення протону (швидкість реакції спадає в порядку, як у реакцій SN1: I > Br > Cl > F).[9]
Якщо бензенове кільце містить інші замісники в орто-положенні до відхідної групи, то вони можуть диригувати селективність реакції:[12]
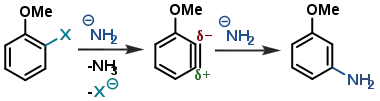
Таким чином, електронодонорні замісники результують сіне-продукт, а електроноакцепторні замісники — іпсо-продукт. Однак, селективність таких реакцій не дуже велика, тому у випадку монозаміщених бензінів зазвичай утворюються обидва регіоізомери.[13]
Механізм SN1
Такий механізм не є характерним для галогеноаренів та арилсульфонатів через низьку стабільність катіону бензену; він має місце лише у виключних випадках.[14] Натомість, найважливішим субстратом для механізму SN1 залишаються діазосполуки (хоча сполуки арилйодонію Ar2I+ теж можуть вступати в реакції заміщення за цим механізмом).[9]
Нуклеофільне ароматичне заміщення за типом SN1 відбувається у два кроки; перший крок — відщеплення відхідної групи — є лімітуючим; загальна швидкість реакції відповідає кінетиці першого порядку[15] і не залежить від концентрації нуклеофілу:[9]

Формування арильних катіонів із діазосполук має певні механістичні особливості:



Вікарне нуклеофільне заміщення (VNS)
Цей особливий тип реакцій SNAr відбуваєтьтся між нітроаренами й аніонами C–H кислот, що містять атом галогену в α-положенні до негативного заряду. В результаті атом водню на ароматичному кільці заміщується нуклеофілом, але відхідна група X- покидає не арен, а нуклеофіл (звідси й назва «вікарне» — тобто, «замісне»). Нітрогрупа у цьому випадку стабілізує комплекс Мейзенгеймера і є основною вимогою для проведення таких реакцій:[17]

Механізм реакції VNS дуже схожий на механізм SNAr; різниця полягає в тому, що утворений комплекс Мейзенгеймера не відщеплює в якості відхідної групи «аніон водню» — натомість, в результаті реакції β-елімінування відщеплюється молекула HX (X є замісником на нуклеофільній сполуці, в даному випадку X = Cl):
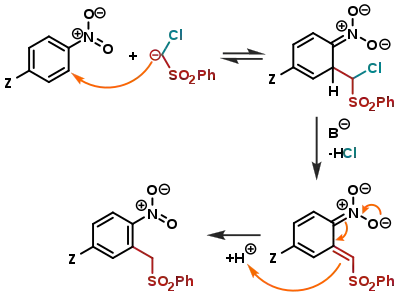
Замісник Z на цій схемі є одним із наведених: Z = F, Cl, Br, I, OCH3, OPh, SCH3, C2H6, tBu, Ph, NMe2, CF3, CN, SO2CH3, COOH.[18] Реакції VNS також ефективно використовуються в синтезі гетероциклів (зокрема, індолів).[19]
Механізм ANRORC
Механізм ANRORC (англ. addition of nucleophile, ring opening and ring closure) є типом реакцій за участю гетероциклічних субстратів, при якому ароматичне кільце спочатку розривається, а потім замикається знов.
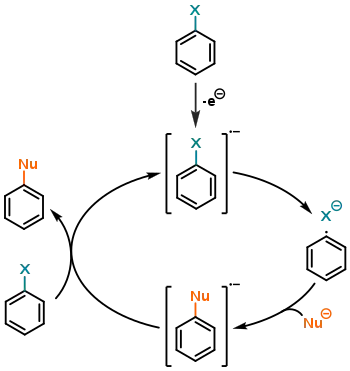
Радикально-нуклеофільне ароматичне заміщення
У цьому типі реакцій, що був відкритий в 1970 році Ф. Буннетом і Дж. Кімом,[20] починається з одноелектронного відновлення (англ. single electron reduction, SER) субстрату. Абревіатура SRN1 означає «заміщення радикально-нуклеофільне одномолекулярне». Найвідомішим прикладом є реакція Зандмеєра.
Реакція SRN1 не потребує активуючих замісників на ароматичному ядрі,[21] однак основною вимогою є наявність радикального ініціатора, що здатен вступати в реакцію SER із субстратом (у випадку реакції Зандмеєра це галогенід міді(I) CuX).
Загальний механізм виглядає наступним чином:
Примітки
- Carey, Francis A., 1937- (1990). Advanced organic chemistry (вид. 3rd ed). New York: Plenum Press. ISBN 978-1-4613-9795-3. OCLC 680545725.
- Vollhardt, K. Peter C.,. Organic chemistry : structure and function (вид. Seventh edition). New York, NY. ISBN 978-1-4641-2027-5. OCLC 866584251.
- Buncel, E.; Norris, A. R.; Russell, K. E. (1968). The interaction of aromatic nitro-compounds with bases. Quarterly Reviews, Chemical Society 22 (2). с. 123. ISSN 0009-2681. doi:10.1039/qr9682200123.
- Strauss, Michael J. (1970-12). Anionic sigma complexes. Chemical Reviews 70 (6). с. 667–712. ISSN 0009-2665. doi:10.1021/cr60268a003.
- Bernasconi, Claude F. (1978-04). Kinetic behavior of short-lived anionic .sigma. complexes. Accounts of Chemical Research 11 (4). с. 147–152. ISSN 0001-4842. doi:10.1021/ar50124a004.
- Briner, G. Peter; Miller, Joseph; Liveris, M.; Lutz, P. G. (1954). The S N mechanism in aromatic compounds. Part VII. Journal of the Chemical Society (Resumed). с. 1265. ISSN 0368-1769. doi:10.1039/jr9540001265.
- Bunnett, J. F.; Garbisch, Edgar W.; Pruitt, Kenneth M. (1957-01). The “Element Effect” as a Criterion of Mechanism in Activated Aromatic Nucleophilic Substitution Reactions1,2. Journal of the American Chemical Society 79 (2). с. 385–391. ISSN 0002-7863. doi:10.1021/ja01559a040.
- Bartoli, Giuseppe; Todesco, Paolo Edgardo (1977-04). Nucleophilic substitution. Linear free energy relations between reactivity and physical properties of leaving groups and substrates. Accounts of Chemical Research 10 (4). с. 125–132. ISSN 0001-4842. doi:10.1021/ar50112a004.
- Smith, Michael B.; March, Jerry (27 грудня 2006). March's Advanced Organic Chemistry. Hoboken, NJ, USA: John Wiley & Sons, Inc. ISBN 978-0-470-08496-0.
- Suwiński, Jerzy; Świerczek, Krzysztof (2001-02). cine - and tele -Substitution reactions. Tetrahedron (англ.) 57 (9). с. 1639–1662. doi:10.1016/S0040-4020(00)01067-X.
- Roberts, John D.; Semenow, Dorothy A.; Simmons, Howard E.; Carlsmith, L. A. (1956-02). The Mechanism of Aminations of Halobenzenes1. Journal of the American Chemical Society 78 (3). с. 601–611. ISSN 0002-7863. doi:10.1021/ja01584a024. Процитовано 1 травня 2020.
- Gilman, Henry; Avakian, Souren (1945-03). Dibenzofuran. XXIII. Rearrangement of Halogen Compounds in Amination by Sodamide1. Journal of the American Chemical Society 67 (3). с. 349–351. ISSN 0002-7863. doi:10.1021/ja01219a001. Процитовано 1 травня 2020.
- Biehl, Edward R.; Nieh, Edward; Hsu, K. C. (1969-11). Substituent effects on the reactivity of arynes. Product distributions as an index of relative reactivities of arynes in methylamine and dimethylamine solvents. The Journal of Organic Chemistry 34 (11). с. 3595–3599. ISSN 0022-3263. doi:10.1021/jo01263a081.
- Himeshima, Yoshio; Kobayashi, Hiroshi; Sonoda, Takaaki (1985-09). A first example of generating aryl cations in the solvolysis of aryl triflates in trifluoroethanol. Journal of the American Chemical Society 107 (18). с. 5286–5288. ISSN 0002-7863. doi:10.1021/ja00304a050.
- Lewis, Edward S.; Miller, Emery B. (1953-01). The Effect of Structure of the Alkyl Group on the Rates of Decomposition of Alkyl Substituted Benzenediazonium Salts1. Journal of the American Chemical Society 75 (2). с. 429–432. ISSN 0002-7863. doi:10.1021/ja01098a050.
- Zollinger, H. (1 січня 1983). Mechanistic investigations on reactions of molecular nitrogen with organic species. Pure and Applied Chemistry (англ.) 55 (2). с. 401–408. ISSN 0033-4545. doi:10.1351/pac198855020401.
- Goliński, Jerzy; Makosza, Mieczysław (1978-01). “Vicarious” nucleophilic substitution of hydrogen in aromatic nitro compounds. Tetrahedron Letters 19 (37). с. 3495–3498. ISSN 0040-4039. doi:10.1016/s0040-4039(00)70555-7.
- Makosza, Mieczyslaw; Winiarski, Jerzy (1987-08). Vicarious nucleophilic substitution of hydrogen. Accounts of Chemical Research (англ.) 20 (8). с. 282–289. ISSN 0001-4842. doi:10.1021/ar00140a003.
- Makosza, M. (1 січня 1997). Synthesis of heterocyclic compounds via vicarious nucleophilic substitution of hydrogen. Pure and Applied Chemistry 69 (3). с. 559–564. ISSN 1365-3075. doi:10.1351/pac199769030559.
- Evidence for a radical mechanism of aromatic «nucleophilic» substitution Joseph F. Bunnett and Jhong Kook Kim J. Am. Chem. Soc.; 1970; 92(25) pp 7463 — 7464. (DOI:10.1021/ja00728a037)
- Rossi, R. A.; Pierini, A. B.; Santiago, A. N. Org. React. 1999, 54, 1. DOI:10.1002/0471264180.or054.01
