Ароматичність
Аромати́чність (англ. Aromaticity) — концепт з області теорії хімічного зв'язку, який застосовується для пояснення особливих властивостей ароматичних сполук. Цей концепт описує феноменальну термодинамічну стабільність сполук з циклічно кон'югованими π-електронами, а також переважання реакцій електрофільного заміщення над реакціями приєднання. Антонімом терміну ароматичності є "антиароматичність" — незвично низька стабільність планарних сполук з циклічно кон'югованими π-електронами (тобто, антиароматичних сполук). Сполуки, що не є ані ароматичними, ані антиароматичними, називають неароматичними.[1]


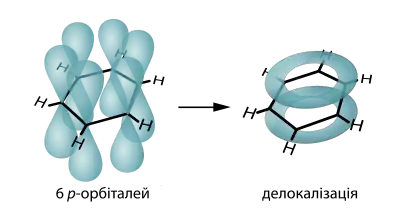
Класичним прикладом ароматичної сполуки незмінно залишається бензен; за своїми властивостями ароматичні сполуки мають бути в певній мірі схожими на нього. Однак, з розвитком теорії хімії критерії ароматичності значно розширились, а з ними все більше різних класів сполук відносили до ароматичних. Також відкривались нові типи ароматичності окрім "класичної" π-ароматичності: гетероароматичність, гомоароматичність, ароматичність Мебіуса, ароматичність Бейрда, сферична ароматичність і т.д.
Широко вживаним методом встановлення ароматичності є спостереження діатропності[2] 1Н в ЯМР спектрах.
Кількісно ступінь ароматичності може бути оцінений за величиною енергії резонансу, або за оцінками енергій відповідних ізодесмічних та гомодесмічних реакцій. Поруч з енергетичними, важливими критеріями є структурні (вирівнювання довжин зв’язків у кільці) та магнітні (наяність діамагнітного кільцевого струму, що проявляється в анізотропії магнітної сприйнятливості).
Виникнення та історія терміну
У 1825 році Майкл Фарадей уперше виділив бензен у чистому вигляді шляхом дистиляції світильного газу, а в 1855 році Август Вільгельм Гофман уперше використав термін "ароматичність"[3] відносно бензену та його гомологів: Гофман помітив, що, на відміну від насичених вуглеводнів, сполуки бензену мають виражені характерні запахи. Окрім того, стала помітна особлива стабільність бензену та його похідних: вони не реагують з галогенами, як це роблять, наприклад, алкени.
У 1865 році Ф.Кекуле запропонував першу структурну формулу бензолу як гексагонального 1,3,5-циклогексатрієна.[4][5]
У 1931 році Еріх Гюккель розробив квантово-механічний підхід для пояснення ароматичності.[6] Цей підхід використовується до сьогодні і називається «метод молекулярних орбіталей Гюккеля» (МОГ); відтепер класичним параметром визначення ароматичності було правило Гюккеля.
У 1936/37 роках Полінг та Лондон запропонували теорію ефектів кільцевого струму.[7][8] У 1956 році Попл описав вплив кільцевого струму на хімічний зсув у ЯМР-спектроскопії,[9] а в 1969/70 роках в якості критерію ароматичності хімічних сполук була описана їхня магнітна сприйнятливість.[10][11]
У 1979 році було введено термін "тривимірна ароматичність",[12] а в 1984 році — σ-ароматичність (на прикладі циклопропану).[13]
У 1996 році Пауль фон Раґ-Шлейєр впровадив NICS (nucleus-independent chemical shift) в якості простого квантово-хімічного методу для вивчення ароматичності.[14]
Критерії ароматичності
Згідно поточного стану науки, існує декілька критеріїв визначення, чи є сполука ароматичною чи антиароматичною. Кожен з них має свої певні недоліки та обмеження, тому надійним методом визначення ароматичності є сукупна перевірка за усіма критеріями.
Правило Гюккеля
Згідно цьому правилу, ароматичними є плоскі циклічні системи з моноциклічно кон'югованими π-електронами (n = 0, 1, 2,..). Антиароматичними є плоскі циклічні системи з моноциклічно кон'югованими π-електронами.[1]
Особливу увагу слід приділяти формулюванню "плоскі циклічні системи"; так, циклооктатетраєн є неароматичною сполукою, бо не є планарним. Це правило добре працює для випадків π-ароматичності, але не може бути застосоване до гомоароматичних сполук. Крім того, ароматичність не є логічною величиною; в ряду фуран–пірол–тіофен–бензен ступінь ароматичності росте зліва направо, хоч усі ці сполуки, згідно правилу Гюккеля, є ароматичними.
Терміни ароматичний та антиароматичний у відповідності до правил Гюккеля і топології орбітального перекривання поширені і на опис стабілізації чи дестабілізації перехідних станів перициклічних реакцій. Такі реакції з антиароматичним перехідним станом йдуть повільніше, якщо взагалі йдуть, ніж ті, що мають ароматичний перехідний стан.
Реактивність
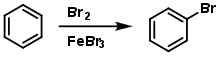
Загалом, ароматичні сполуки не вступають в реакції приєднання; так, бензен не знебарвлює бромну воду. Замість цього він вступає в реакцію електрофільного заміщення в присутності кислоти Льюїса (наприклад, ферум(III)боміду):
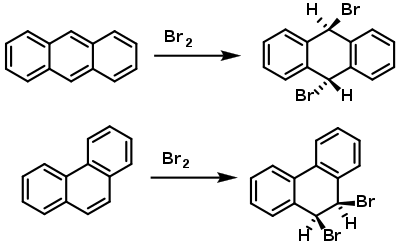
Однак, поліциклічні ароматичні сполуки (антрацен, фенантрен тощо) не відповідають цьому правилу, хоч і виконують правило Гюккеля. При взаємодії з галогенами вони вступають в реакції приєднання (для трициклічних сполук — по центральному кільцю):[1]
Згадані раніше п'ятичленні гетероцикли, виконуючи правило Гюккеля, водночас здатні брати участь у реакціях Дільса–Альдера.[15]
Структура
Враховуючи моноциклічну кон'югацію π-орбіталей в ароматичних сполуках, довжини зв'язків між атомами Карбону мають бути (майже) однаковими. Ступінь схожості довжин зв'язків визначає ступінь ароматичності сполуки. Однак, цей критерій не можна використовувати ізольовано від інших: як показано нижче, з трьох наведених молекул кадаверин має майже однакові С–С зв'язки, хоча не виконує правило Гюккеля.[16]
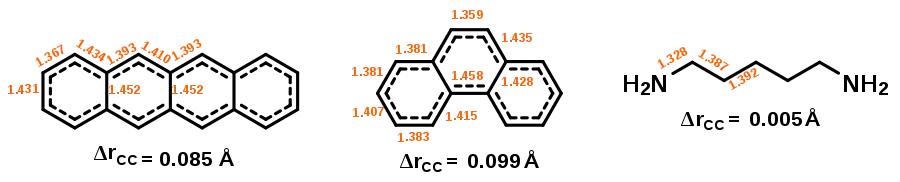
Термохімія
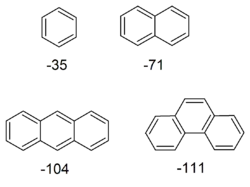
Інше спостереження стосовно ароматичних сполук полягає в тому, що бензен, на відміну від ненасичених сполук, слабо підвержений гідрогенізації. Термохімічні дослідження підтвердили, що теплота, яка виділяється при гідрогенізації бензену, нижча в порівнянні з гіпотетичним неароматичним "циклогексатрієном" на 35 ккал/моль.[17] Ця різниця називається енергія ароматичної стабілізації (ЕАС; англ. Aromatic stabilization energy, ASE). В літературі зустрічаються певні розбіжності щодо знаку перед величиною ЕАС, але в цій статті "стабілізація" буде позначатись негативним знаком, що відповідає негативній , тобто виділенню теплоти.

Альтернативою термохімічним вимірам можуть бути квантово-механічні розрахунки.[18] Найпоширенішим методом є медод ізодесмічних реакцій (у цьому випадку, оскільки має враховуватися зміна гібридизації атомів Карбону, вони називаються гомодесмотичними реакціями). Альтернативно, Шлейєр et al. запропонували використовувати для розрахунків реакції ізомеризації;[19] отримані енергії ізомеризаційної стабілізації (англ. isomerization stabilization energies) добре узгоджуються з результатами, що були отримані іншими методами.
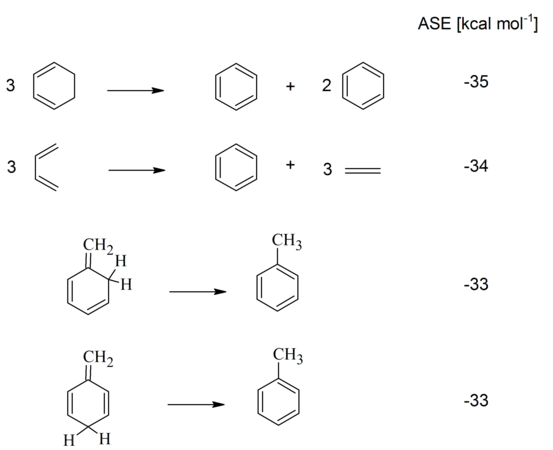
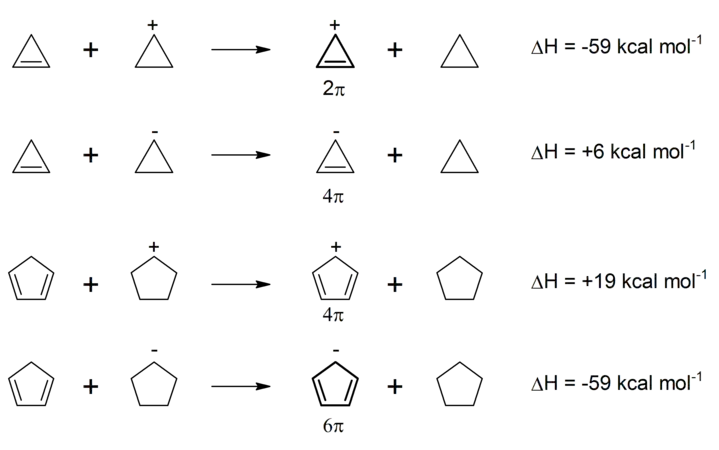
ЯМР-спектроскопія
У сполуках з циклічно кон'югованими π-орбіталями делокалізація π-електронів спричиняє появу кільцевого струму, який, згідно закону Біо–Савара–Лапласа (або ж Правилу правої руки), утворює локальне магнітне поле молекули. При розташуванні такої молекули в сильному зовнішньому магнітному полі (наприклад, у ЯМР-спектрометрі) лінії локального поля можуть вирівнятись або паралельно, або антипаралельно лініям зовнішнього поля:
- При паралельній орієнтації атоми Гідрогену зазнають додаткового екранування, їхній хім. зсув зменшується; такий ефект в ЯМР називають паратропним, а кільцевий струм у циклічній молекулі — "парамагнетичним кільцевим струмом".
- При антипаралельній орієнтації атоми Гідрогену деекранізуються локальним полем, їхній хім. зсув збільшується; такий ефект в ЯМР називають діатропним, а кільцевий струм отримує назву "діамагнетичний кільцевий струм".[22]
Наявність паратропного ефекту свідчить про антиароматичність кільця, а діатропний ефект свідчить про його ароматичність:
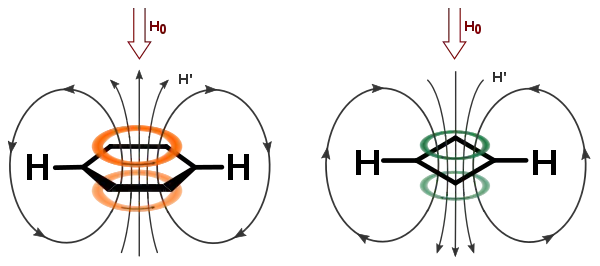
Таке явище можна спрощено запам'ятати наступним чином: при заповненні МО бензену та циклобутадієну (див. Цикл Фроста) всі π-електрони бензену будуть у спареному стані (що свідчить про його діамагнітні властивості); натомість, відповідно до цієї моделі, циклобутадієн є бірадикалом, а тому є парамагнетиком.
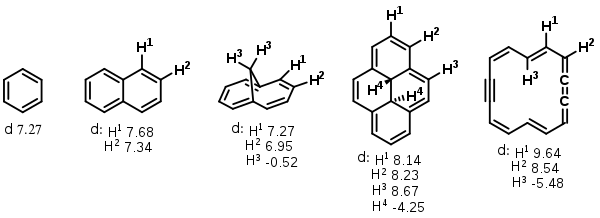
Нижче наведені хімічні зсуви атомів Гідрогену в 1Н ЯМР для різних ароматичних та неароматичних ануленів:[22]
| Ароматичні системи | Антиароматичні системи | ||||
|---|---|---|---|---|---|
| Анулен
або його іон |
Зовнішні
протони |
Внутрішні
протони |
Анулен
або його іон |
Зовнішні
протони |
Внутрішні
протони |
| [6] | 7.37 | – | [8] | 5.68 | - |
| [14] | 7.6 | 0 | [12] | 5.91 | 7.86 |
| [18] | 9.28 | -2.99 | [16] | 5.40 | 10.43 |
| [22] | 8.50 – 9.65 | 0.4 – -1.2 | [20] | 4.1 – 6.6 | 10.9 – 13.9 |
| [12]-2 | 6.23, 6.98 | -4.6 | [18]-2 | 1.13 | 28.1, 29.5 |
| [16]+2 | 7.45, 8.83 | -8.17 | |||
Крім 1Н ЯМР, 7Li ЯМР також використовуються для вивчення ароматичних властивостей, зокрема аніонних π-систем, оскільки катіони Li+, як правило, розташовуються у центральному положенні над π-системою:[20]
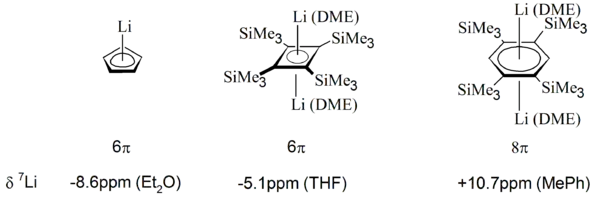
Nucleus-Independent Chemical Shift (NICS)
З розвитком методів квантово-хімічних розрахунків стало можливим обчислити хімічні зсуви умовного протону в будь-якій точці простору і, таким чином, оцінити вплив кільцевого струму незалежно від того, з яких атомів побудований. Цей метод отримав назву Nucleus Independent Chemical Shift (NICS).[20] Для дослідження ароматичних сполук величини NICS зазвичай обчислюються в геометричному центрі групи важких атомів (тобто будь-яких атомів важче за Гідроген і Гелій). Величини NICS нормовані таким чином, щоб узгоджуватись із хімічними зсувами протонів у 1Н ЯМР. Тобто, негативні значення всередині циклу вказують на ароматичність, а позитивні — на антиароматичність.
Незважаючи на те, що для (анти)ароматичних молекул вплив кільцевого струму є головним фактором, що визначає величину NICS, локалізовані зв’язки та самотні електронні пари також мають певний вплив на короткій відстані, тому є заважаючими факторами. Для усунення цього впливу, величину NICS можна обчислити у площині (анти)ароматичного кільця. Більш складні методи також дозволяють розраховувати внески до значення NICS окремих σ- й π-зв’язків, а також окремих молекулярних орбіталей.
Типи ароматичності
Існує три типи ароматичності: Ароматичність Гюккеля, ароматичність Мебіуса та ароматичність Бейрда.
Ароматичність Гюккеля
Формально описується правилом Гюккеля. Може бути поділена на наступні види:
- π-Ароматичність: Класичний випадок ароматичності; циклічна делокалізація відбувається шляхом кон'югації π-орбіталей по замкнутому периметру, утвореному σ-зв’язками.
- Гетероароматичність: Випадок π-ароматичності, коли окремі атоми Карбону замінено гетероатомами (див. Гетероциклічні сполуки).
- σ-Ароматичність: Для малих кілець (особливо циклопропанів) спостерігаються енергетична стабілізація та магнітні властивості, притаманні π-ароматичним сполукам. Прикладом σ-ароматичності серед неорганічних сполук є катіон Н3+.
- Гомоароматичність: Зустрічається у випадках, коли класична π-ароматичність розривається в одній точці, наприклад, метиленовою групою; за виконанням певних геометричних умов, π-делокалізація може підтримуватись через простір. Гарним прикладом цього феномену є катіон гомотропілію. Якщо кон'югацію перервано в декількох місцях, то в окремих випадках можна говорити про ароматичність біс-гомо, тріс-гомо тощо.[24]
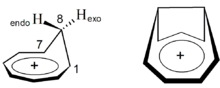 Катіони гомотропілію й біс-гомортопілію.[23]
Катіони гомотропілію й біс-гомортопілію.[23] - Сферична ароматичність (просторова ароматичність, 3D ароматичність): Ароматичність у трьох вимірах існує у випадках кластерних сполук (напр., борани) або великих непланарних молекул з великою кількістю ненасичених зв'язків (фулерени).[25]
Ароматичність Мебіуса
Циклічно кон'юговані молекули, що мають певне число інверсій фази π-орбіталей (подібно стрічці Мебіуса) є ароматичними, якщо в делокалізації беруть участь π-електронів (n = 0, 1, 2,..).
Ароматичність Бейрда
В 1972 р. Н. Колін Бейрд опублікував квантово-механічні розрахунки, що прогнозували ароматичність найнижчого триплетного стану ануленів, які мають π-електронів.[26] Таким чином, катіон Cp+ є ароматичним при збудженні та інтеркомбінаційній конверсії із синглетного у триплетний стан, що було доведено за допомогою ЕПР-спектроскопії на прикладі молекул [Ph5Cp]+[27] і [Cl5Cp]+.[28]
Примітки
- Anslyn, Eric V., 1960- (2006). Modern physical organic chemistry. Sausalito, CA: University Science. ISBN 1-891389-31-9. OCLC 55600610.
- Дiатропна сполука - ароматична сполука яка здатна утримувати iндукований кiльцевий струм, наявність якого встановлюється за допомогою хімiчних зсувiв у спектрах ЯМР.
- I. On insolinic acid. Proceedings of the Royal Society of London (англ.) 8. 31 грудня 1857. с. 1–3. ISSN 0370-1662. doi:10.1098/rspl.1856.0002.
- Kekulé, F. A. (1865). Sur la constitution des substances aromatiques. Bulletin de la Société Chimique de Paris 3: 98–110.
- Kekulé, F. A. (1866). Untersuchungen über aromatische Verbindungen (Investigations of aromatic compounds). Liebigs Annalen der Chemie und Pharmacie 137 (2): 129–36. doi:10.1002/jlac.18661370202.
- E. Hückel, Zeitschrift für Physik, 70, 204 (1931); 72, 310 (1931); 76, 628 (1932); 83, 632 (1933).
- L. Pauling: The Diamagnetic Anisotropy of Aromatic Molecules. In: J. Chem. Phys., 1936, 4, S. 673–677.
- F. London: Théorie quantique des courants interatomiques dans les combinaisons aromatiques. In: J. Phys. Radium 1937, 8, S. 397–409.
- J. A. Pople: Proton Magnetic Resonance of Hydrocarbons. In: J. Chem. Phys., 1956, 24, S. 1111.
- Hyp J. Dauben, James Dennis. Wilson, John L. Laity: Diamagnetic susceptibility exaltation as a criterion of aromaticity. In: Journal of the American Chemical Society. 90, 1968, S. 811, doi:10.1021/ja01005a059.
- R. C. Benson, W. H. Flygare: Molecular Zeeman effect of cyclopentadiene and isoprene and comparison of the magnetic susceptibility anisotropies. In: J. Am. Chem. Soc., 1970, 92, S. 7523–7529.
- J. Aihara: Three-dimensional aromaticity of polyhedral boranes. In: J. Am. Chem. Soc., 1978, 100, S. 3339–3342.
- Michael J. S. Dewar: Chemical implications of σ conjugation. In: Journal of the American Chemical Society. 106, 1984, S. 669, doi:10.1021/ja00315a036.
- P. v. Ragué-Schleyer, C. Maerker, A. Dransfeld, H. Jiao, N. J. R. v. E. Hommes: Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. In: J. Am. Chem. Soc., 1996, 118, S. 6317–6318.
- Sankararaman, S. (Sethuraman), 1957- (2005). Pericyclic reactions : a textbook : reactions, applications, and theory. Weinheim: Wiley-VCH. ISBN 3-527-31439-3. OCLC 61439936.
- von Schleyer, P. Rague; Jiao, Haijun (1 січня 1996). What is aromaticity?. Pure and Applied Chemistry 68 (2). с. 209–218. ISSN 1365-3075. doi:10.1351/pac199668020209.
- J. D. Roberts, M. C. Caserio: Basic Principles of Organic Chemistry, W. A. Benjamin Inc., New York, Amsterdam, 1965.
- Michał Ksawery Cyrański: Energetic Aspects of Cyclic Pi-Electron Delocalization: Evaluation of the Methods of Estimating Aromatic Stabilization Energies. In: Chem. Rev. 2005, 105, S. 3773–3811, doi:10.1021/cr0300845.
- Schleyer et al. in Chem. Rev., 2005, 105, S. 3842–3888 und dort zitierte Literatur.
- Z. Chen, C.S. Wannere, C. Corminboeuf, R. Puchta, P. v. R. Schleyer: Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. In: Chemical Reviews, 2005, 105, S. 3842, DOI:10.1021/cr030088+
- Kenneth B. Wiberg: Antiaromaticity in Monocyclic Conjugated Carbon Rings. In: Chemical Reviews. 101, 2001, S. 1317, doi:10.1021/cr990367q.
- Minkin, V. I. (Vladimir Isaakovich); Simkin, B. I︠A︡. (Boris I︠A︡kovlevich) (1994). Aromaticity and antiaromaticity : electronic and structural aspects. New York: Wiley. ISBN 0-471-59382-6. OCLC 27974900.
- D. Cremer, P. Svensson, E. Kraka, Z. Konkoli, P. Ahlberg: Exploration of the Potential Energy Surface of C9H9+ by ab Initio Methods. 2. Is the 1,4-Bishomotropylium Cation a Bishomoaromatic Prototype?. In: J. Am. Chem. Soc., 1993, 115, S. 7457–7464.
- R. V. Williams: Homoaromaticity. In: Chem. Rev., 2001, 101, S. 1185–1204.
- Z. Chen, R. B. King: Spherical Aromaticity: Recent Work on Fullerenes, Polyhedral Boranes, and Related Structures. In: Chem. Rev., 2005, 105, S. 3613–3642.
- Baird, N. Colin (1972-07). Quantum organic photochemistry. II. Resonance and aromaticity in the lowest 3.pi..pi.* state of cyclic hydrocarbons. Journal of the American Chemical Society (англ.) 94 (14). с. 4941–4948. ISSN 0002-7863. doi:10.1021/ja00769a025.
- Yager, William A. (1963-07). A Stable Triplet State of Pentaphenylcyclopentadienyl Cation. Journal of the American Chemical Society 85 (13). с. 2033–2034. ISSN 0002-7863. doi:10.1021/ja00896a042.
- Breslow, Ronald.; Hill, Roger.; Wasserman, E. (1964-12). Pentachlorocyclopentadienyl Cation, a Ground-State Triplet. Journal of the American Chemical Society 86 (23). с. 5349–5350. ISSN 0002-7863. doi:10.1021/ja01077a072.
