Спадковий неполіпозний колоректальний рак
Спадковий неполіпозний колоректальний рак (HNPCC) або Синдром Лінча — аутосомно-домінантна генетична патологія, пов’язана з високим ризиком виникнення раку товстої кишки [2], а також інших видів раку: раку ендометрію (другий за поширеністю), раку яєчника, раку шлунка, раку тонкого кишечника, раку гепатобіліарного тракту, раку верхніх сечових шляхів, раку головного мозку та раку шкіри. Підвищений ризик цих новоутворень обумовлений успадкованими мутаціями, які уражають механізм репарації помилково спарених нуклеотидів (MMR). Це один з видів онкологічного синдрому.
| Спадковий неполіпозний колоректальний рак | |
|---|---|
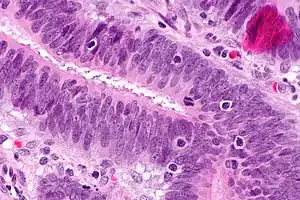 Мікрофотографія демонструє лімфоцити, що інфільтрують пухлину (при колоректальному раку), знахідка мікросатітної нестабільності пухлини, що характерно для синдрому Лінча. фарбування гематоксилін-еозином. Мікрофотографія демонструє лімфоцити, що інфільтрують пухлину (при колоректальному раку), знахідка мікросатітної нестабільності пухлини, що характерно для синдрому Лінча. фарбування гематоксилін-еозином. | |
| Інші назви | Синдром Лінча[1] |
| Спеціальність | онкологія |
| Класифікація та зовнішні ресурси | |
| МКХ-10 | C18 і C20 |
| OMIM | 120435 |
| DiseasesDB | 5812 |
| MeSH | D003123 |
Ознаки та симптоми
Ризик раку
Ризик протягом житта та середній вік при діагностиці раку, асоційованого із синдромом Лінча [3]
| Тип раку | Ризик протягом життя (%) | Середній вік при діагностиці (роки) |
| Колоректальний | 52-58 | 44-61 |
| Ендометріальний | 25-60 | 48-62 |
| Шлунковий | 6-13 | 56 |
| Яєчниковий | 4-12 | 42.5 |
На додаток до типів раку, вказаних у таблиці вище, вважається, що синдром Лінча також сприяє підвищенню ризику раку тонкого кишечника, раку підшлункової залози, раку сечоводу/ниркової миски, раку жовчовивідних шляхів, пухлин головного мозку та пухлин сальних залоз. [3] Підвищений ризик раку передміхурової залози та раку молочної залози також пов’язаний із синдромом Лінча, хоча цей взаємозв'язок не зовсім зрозумілий.
Дві третини випадків раку товстої кишки трапляються в проксимальному відділі. Середній вік на момент діагнозу колоректального раку становить 44 роки для членів сімей, які відповідають Амстердамським критеріям (див. далі). Середній вік діагностики раку ендометрію становить близько 46 років. Серед жінок із HNPCC, у яких є рак товстої кишки та ендометрію, у приблизно половини рак ендометрію виникає першим, що робить його найпоширенішим первинним раком при синдромі Лінча. [4] При HNPCC середній вік діагностики раку шлунка становить 56 років, аденокарцинома кишкового типу є найчастішим варіантом. Рак яєчників, асоційований з HNPCC, має середній вік діагнозу 42,5 років; приблизно у 30 % він діагностується у віці до 40 років.
Велике спостережне дослідження (3 119 пацієнтів; середнє спостереження протягом 24 років) виявило значну різницю частоти раку в залежності від мутації. [5] До 75 років ризик колоректального раку, раку ендометрія, раку яєчників, раку верхніх відділів шлунково-кишкового тракту (шлунка, дванадцятипалої кишки, жовчної протоки або підшлункової залози), раку сечовивідних шляхів, раку передміхурової залози та пухлин мозку були такими: для мутації MLH1 ризик був - 46 %, 43 %, 10 %, 21 %, 8 %, 17 % та 1 % відповідно; для мутацій MSH2 ризики становили 57 %, 17 %, 10 %, 25 %, 32 % та 5 % відповідно; для мутацій MSH6 ризики становили 15 %, 46 %, 13 %, 7 %, 11 %, 18 % та 1 % відповідно.
| Мутований ген | Ризик раку яєчників, % | Ризик раку ендометрію, % |
|---|---|---|
| MLH1 | 4-24 | 25-60 |
| MSH2 / EPCAM | 4-24 | 25-60 |
| MSH6 | 1-11 | 16-26 |
| PMS2 | 6 (комбінований ризик) | 15 |
Генетика
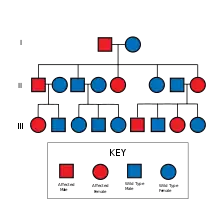
HNPCC успадковується аутосомно-домінантно. [7] Відмітною ознакою HNPCC є несправний механізм репарації помилково спарених нуклеотидів, що призводить до мікросателітної нестабільності (MSI), також відомої як MSI-H (Н - "high, високий"). MSI ідентифікується в зразках пухлин при гістологічному дослідженні. У більшості випадків це призводить до зміни довжини тандемних повторів нуклеїнових основ цитозину та аденіну (послідовність: CACACACACA. . . ). [8]
Чотири основні гени, що залучені у розвиток HNPCC, зазвичай кодують білки, які для функціонування утворюють димери:
- Білок MLH1 димеризується з білком PMS2, утворюючи MutLα, який координує зв'язування інших білків, що беруть участь у репарації помилково спарених нуклеотидів, як геліказа ДНК; білок, зв'язуючий одноланцюгову ДНК (RPA) та ДНК полімерази . [9] [10]
- Білок MSH2 димеризується з білком MSH6, який ідентифікує помилки спарювання нуклеотидів за допомогою моделі ковзаючого затискача, білок для сканування на наявність помилок. [11] [12]
Порушення будь-якого гена білкового димеру погіршує функцію білка. [13] Ці 4 гена беруть участь у виправленні помилок ДНК, тому їх дисфункція може призвести до неможливості виправити помилки реплікації ДНК та викликати HNPCC. [14] Відомо, що HNPCC асоціюється з іншими мутаціями генів, які беруть участь у репарації помилково спарених нуклеотидів :
| OMIM | Гени, залучені у HNPCC | Частота мутацій у сім'ях HNPCC | Локус | Перша публікація |
|---|---|---|---|---|
| HNPCC1 | MSH2 / EPCAM | приблизно 60 % | 2p22 | Фішель 1993 р. [15] |
| HNPCC2 | MLH1 | приблизно 30 % | 3p21 | Пападопулос 1994 [16] |
| HNPCC5 | MSH6 | 7-10 % | 2п16 | Міякі 1997 [17] |
| HNPCC4 | PMS2 | відносно нечасті | 7p22 | Nicolaides 1994 |
| HNPCC3 | PMS1 | одиничні випадки [18] | 2q31-q33 | Nicolaides 1994 |
| HNPCC6 | TGFBR2 | одиничні випадки [19] | 3p22 | |
| HNPCC7 | MLH3 | дискутується [20] | 14q24.3 | |
Люди з мутацією MSH6, швидше за все, мають негативні Амстердамські критерії II. [21] Прояви мутацій MSH6 дещо відрізняються від мутацій MLH1 та MSH2, для опису цього стану був використаний термін "синдром MSH6". [22] В одному дослідженні Рекомендації Бетесда були більш чутливими, ніж Амстердамські критерії, щодо виявлення проявів цих мутацій. [23]
До 39 % сімей з мутаціями генів, асоційованих з HNPCC, не відповідають Амстердамським критеріям. Тому сім'ї, у яких виявлено шкідливу мутацію генів HNPCC, слід вважати такими, що мають HNPCC незалежно від сімейного анамнезу. Це також означає, що Амстердамські критерії не здатні виявити багатьох людей, які мають ризик синдрому Лінча. Удосконалення критеріїв скринінгу є активною галуззю досліджень, детально описаною в розділі "Стратегії скринінгу" цієї статті.
Більшість людей із HNPCC успадковують патологію від батька. Однак через неповну пенетрантність, мінливий вік діагностики раку, зниження ризику раку або ранню смерть не всі люди з мутацією гена HNPCC мають батьків, у яких був рак. У деяких людей HNPCC виникає de-novo, без успадковування мутації. Ці пацієнти часто виявляються лише після розвитку раку товстої кишки у ранньому віці. Батьки з HNPCC мають 50 % шанс передати генетичну мутацію кожній дитині. Важливо також зазначити, що шкідливих мутацій лише в одному з генів MMR недостатньо для того, щоб викликати рак, для цього потрібні подальші мутації в інших генах-супресорах. [24]
Діагностика
Діагноз синдрому Лінча встановлюється людям із мутацією ДНК статевих клітин в одному з генів MMR (MLH1, MSH2, MSH6 та PMS2) або у гені EPCAM, виявленими генетичним тестуванням. [25] Кандидати на генетичне тестування статевих клітин можуть бути визначені за такими клінічними критеріями, як Амстердамські клінічні критерії та Рекомендації Бетесда, або за допомогою аналізу пухлин методом імуногістохімії (IHC), або тестуванням на мікросателітну нестабільність (MSI). Генетичне тестування є комерційно доступним і полягає у аналізі крові.
Імуногістохімія
Імуногістохімія (IHC) - це метод, який може бути використаний для виявлення аномальної експресії білків MMR у пухлинах, пов'язаних із синдромом Лінча. Хоча вона не діагностує синдром Лінча, вона може відігравати певну роль у виявленні людей, яким потрібно провести тестування статевих клітин. [26] Використовують два методи тестування: за віком та тестування всіх людей. [27] Наразі немає згоди щодо того, який метод скринінгу слід використовувати. Тестування за віком було запропоновано частково завдяки аналізу витрат та вигод, тоді як тестування всіх пацієнтів, хворих на колоректальний рак, гарантує, що люди з синдромом Лінча не будуть пропущені.
Мікросателітна нестабільність
Мутації в системі репарації помилково спарених нуклеотидів можуть призвести до копіювання ділянок всередині ДНК, які містять повторювані структури двох-трьох нуклеотидів (мікросателітів), інакше відомих як мікросателітна нестабільність (MSI). [28] MSI ідентифікують шляхом екстракції ДНК зі зразка пухлинної тканини, та зі зразка нормальної тканини з подальшим ПЛР-аналізом мікросателітних ділянок. MSI-аналіз може бути використаний для виявлення людей, які можуть мати синдром Лінча, та спрямування їх на подальше тестування.
Класифікація
За гістопатологічними критеріями можна виділити три основні групи раку з високою мікросателітною нестабільністю (MSI-H):
- правобічний поганодиференційований рак
- правобічний муцинозний рак
- аденокарциноми будь-якої локалізації, що мають будь-яку вимірювану концентрацію внутрішньоепітеліальних лімфоцитів (TIL)
Крім того, HNPCC можна розділити на синдром Лінча I (сімейний рак товстої кишки) та синдром Лінча II (HNPCC, пов’язаний з іншими раковими захворюваннями шлунково-кишкового тракту або репродуктивної системи ). [29]
Профілактика
Після публікації про відсутність результатів у рандомізованому контрольованому дослідженні аспірину (ацетилсаліцилова кислота - АСК) для запобігання колоректальних новоутворень при синдромі Лінча, [30] Бьорн та його колеги повідомили нові дані про результати більш тривалого періоду спостереження, ніж у першій статті. Ці нові дані показали зниження захворюваності із синдромом Лінча, які отримували високу дозу аспірину щонайменше чотири роки при задовільному ризику ускладнень. [31] Ці результати широко були розголошені у ЗМІ; майбутні дослідження присвячені питанню зниження дози (для зменшення ризику кровотеч, пов’язаного з високою дозою АСК).
Скринінг
Сім'ям, які відповідають Амстердамським критеріям до появи раку товстої кишки рекомендується генетична консультація та генетичне тестування.
Для скринінгу раку яєчників та ендометрію рекомендується щорічне трансвагінальне УЗД з біопсією ендометрію. [6]
Амстердамські критерії
Нижче наведено Амстердамські критерії для визначення високоризикових кандидатів для молекулярно-генетичного тестування: [32]
Амстердамські критерії I (усі пункти повинні бути заповнені): були опубліковані в 1990 році; проте, вони вважалися недостатньо чутливими. [33]
- Три або більше членів сім'ї з підтвердженим діагнозом колоректального раку, один з яких є родичем першого ступеню (батьком, дитиною, рідним братом) двох інших
- Хвороба у двох послідовних поколінь
- Один або кілька раків товстої кишки, діагностованих у віці до 50 років
- Сімейний аденоматозний поліпоз (FAP) виключений
Амстердамські критерії II були розроблені в 1999 році та покращили діагностичну чутливість, через додання раку ендометрію, тонкої кишки, сечоводу та ниркової миски.[34]
Амстердамські критерії ІI (всі пукти повинні бути заповнені): [34]
- Три або більше членів сім’ї з HNPCC-асоційованим раком, один з яких є родичем першого ступеня двох інших
- Ураження двох послідовних поколінь
- Одне або декілька HNPCC-асоційованих ракових захворювань, діагностованих у віці до 50 років
- Сімейний аденоматозний поліпоз (FAP) виключений
Критерії Бетесда (Bethesda) були розроблені Національним інститутом раку в 1997 році, і оновлені в 2004 році для виявлення осіб, які потребують подальшого тестування методом визначення мікросателітної нестабільності (MSI). На відміну від Амстердамських критеріїв, переглянуті Рекомендації Бетесда на додаток до клінічної інформації використовують патоморфологічні дані. [33] [34]
Переглянуті Рекомендації Бетесда:
Якщо людина відповідає будь-якому одному з 5 критеріїв, пухлина (и) у людини повинна бути протестована на MSI: [33]
1. Колоректальний рак, діагностований у віці до 50 років
2. Наявність синхронних або метахронних колоректальних або інших ракових захворювань, пов'язаних з синдромом Лінча (наприклад, рак ендометрію, яєчників, шлунка, тонкої кишки, підшлункової залози, жовчовивідних шляхів, сечоводу, ниркової миски, мозку, сальних залоз, кератоакантоми )
3. Колоректальний рак з патоморфологією MSI-H у віці до 60 років
4. Колоректальний рак, діагностований у людини з одним або декількома родичами першого ступеня у яких з колоректальний рак або інша пухлина, асоційована з синдромом Лінча, було діагностовано у віці до 50 років
5. Особа з колоректальним раком та двома або більше родичами першого чи другого ступеня з колоректальним раком або іншим раком, асоційованим з синдромом Лінча, діагностованими у будь-якому віці [33] [34]
Важливо зауважити, що клінічні критерії можуть бути важкими для використання на практиці, їх ізольоване застосування може призвести до отримання хибнонегативних результатів діагностики у 12 - 68 % випадків синдрому Лінча. [33]
Хірургічна профілактика
Профілактична гістеректомія та сальпінгооофоректомія (видалення матки, маткових труб та яєчників для запобігання розвитку раку) можуть проводитися до появи раку яєчників або ендометрію. [6]
Лікування
Операція залишається лікуванням вибору для HNPCC. Продовжується дискусія щодо користі ад'ювантної терапії на основі 5-фторурацилу для колоректальних пухлин, пов’язаних з HNPCC, зокрема, на I та II стадіях. [35]
Епідеміологія
У США щороку діагностується близько 160 000 нових випадків колоректального раку. Спадковий неполіпозний колоректальний рак є причиною приблизно від 2 до 7 % від усіх цих випадків. Середній вік діагностики раку у пацієнтів із цим синдромом становить 44 роки, порівняно з 64 роками у людей без синдрому.
Термінологія
Генрі Т. Лінч, професор медицини Медичного центру Крейтонського університету, описав синдром у 1966 році. [37] У своїй пілотній роботі він характеризував хворобу як "синдром сімейного раку". Термін "синдром Лінча" був введений у 1984 р. іншими авторами. У 1985 році Лінч назвав хворобу "спадковий неполіпозний колоректальний рак". Відтоді обидва терміни використовувались взаємозамінно. Нещодавній прогрес у розумінні генетики захворювання призвів до того, що термін HNPCC втрачає популярність. [38]
У деяких джерелах термін "синдром Лінча" використовується, коли мутація, що веде до дефект репарації помилково спарених нуклеотидів є відомою. Термін "сімейний колоректальний рак типу Х", застосовується, коли пацієнт відповідає амстердамським критеріям, але мутація не визначена. [39] Можливо, що сім'ї "типу X" мають нижчу загальну інцидентність раку і нижчий ризик виникнення неколоректального раку, ніж сім'ї з документально підтвердженим дефіцитом репарації помилково спарених нуклеотидів. [40] Близько 35 % людей, які відповідають Амстердамським критеріям, не мають мутації відповідних генів. [41]
Ускладнюючим питанням є наявність альтернативного набору критеріїв, відомих як "Рекомендації Бетесда". [42] [43] [44]
Суспільство
Існує низка неприбуткових організацій, що надають інформацію та підтримку пацієнтам, такі як Lynch Syndrome International, Lynch Syndrome UK [45] та Bowel Cancer UK. [46] У США є Національний день застереження щодо синдрому Лінча - 22 березня. [47]
Примітки
- Lynch syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. rarediseases.info.nih.gov. Процитовано 4 жовтня 2019.
- Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, Bandipalliam P, Stoffel EM, Gruber SB, Syngal S (October 2009). Risk of pancreatic cancer in families with Lynch syndrome. JAMA 302 (16): 1790–5. PMC 4091624. PMID 19861671. doi:10.1001/jama.2009.1529.
- Lynch Syndrome. DynaMed. 22 лютого 2019. Процитовано 18 листопада 2019.
- Hoffman, Barbara L. (2012). Chapter 33: Endometrial Cancer. Williams Gynecology (вид. 2nd). New York: McGraw-Hill Medical. ISBN 978-0071716727.
- Møller P, Seppälä TT, Bernstein I, Holinski-Feder E, Sala P, Gareth Evans D, Lindblom A, Macrae F, Blanco I, Sijmons RH, Jeffries J, Vasen HF, Burn J, Nakken S, Hovig E, Rødland EA, Tharmaratnam K, de Vos Tot Nederveen Cappel WH, Hill J, Wijnen JT, Jenkins MA, Green K, Lalloo F, Sunde L, Mints M, Bertario L, Pineda M, Navarro M, Morak M, Renkonen-Sinisalo L, Valentin MD, Frayling IM, Plazzer JP, Pylvanainen K, Genuardi M, Mecklin JP, Moeslein G, Sampson JR, Capella G (July 2018). path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut 67 (7): 1306–1316. PMC 6031262. PMID 28754778. doi:10.1136/gutjnl-2017-314057.
- Ring KL, Garcia C, Thomas MH, Modesitt SC (November 2017). Current and future role of genetic screening in gynecologic malignancies. American Journal of Obstetrics and Gynecology 217 (5): 512–521. PMID 28411145. doi:10.1016/j.ajog.2017.04.011.
- Lynch Syndrome. Genetics Home Reference.
- Oki E, Oda S, Maehara Y, Sugimachi K (March 1999). Mutated gene-specific phenotypes of dinucleotide repeat instability in human colorectal carcinoma cell lines deficient in DNA mismatch repair. Oncogene 18 (12): 2143–7. PMID 10321739. doi:10.1038/sj.onc.1202583.
- Yokoyama, Takanori; Takehara, Kazuhiro; Sugimoto, Nao; Kaneko, Keika; Fujimoto, Etsuko; Okazawa-Sakai, Mika; Okame, Shinichi; Shiroyama, Yuko та ін. (December 2018). Lynch syndrome-associated endometrial carcinoma with MLH1 germline mutation and MLH1 promoter hypermethylation: a case report and literature review. BMC Cancer 18 (1): 576. ISSN 1471-2407. PMC 5963021. PMID 29783979. doi:10.1186/s12885-018-4489-0.
- Peltomäki, Päivi (15 березня 2003). Role of DNA mismatch repair defects in the pathogenesis of human cancer. Journal of Clinical Oncology 21 (6): 1174–1179. ISSN 0732-183X. PMID 12637487. doi:10.1200/JCO.2003.04.060.
- Tamura, Kazuo; Kaneda, Motohide; Futagawa, Mashu; Takeshita, Miho; Kim, Sanghyuk; Nakama, Mina; Kawashita, Norihito; Tatsumi-Miyajima, Junko (September 2019). Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome. International Journal of Clinical Oncology 24 (9): 999–1011. ISSN 1341-9625. PMID 31273487. doi:10.1007/s10147-019-01494-y.
- Fishel, R.; Lescoe, M. K.; Rao, M. R.; Copeland, N. G.; Jenkins, N. A.; Garber, J.; Kane, M.; Kolodner, R. (3 грудня 1993). The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 75 (5): 1027–1038. ISSN 0092-8674. PMID 8252616. doi:10.1016/0092-8674(93)90546-3.
- Yurgelun, Matthew B.; Hampel, Heather (23 травня 2018). Recent Advances in Lynch Syndrome: Diagnosis, Treatment, and Cancer Prevention. American Society of Clinical Oncology Educational Book. American Society of Clinical Oncology. Annual Meeting 38 (38): 101–109. ISSN 1548-8756. PMID 30231390. doi:10.1200/EDBK_208341.
- Le, Stephanie; Ansari, Umer; Mumtaz, Aisha; Malik, Kunal; Patel, Parth; Doyle, Amanda; Khachemoune, Amor (15 листопада 2017). Lynch Syndrome and Muir-Torre Syndrome: An update and review on the genetics, epidemiology, and management of two related disorders. Dermatology Online Journal 23 (11). ISSN 1087-2108. PMID 29447627.
- Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R (December 1993). The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 75 (5): 1027–38. PMID 8252616. doi:10.1016/0092-8674(93)90546-3.
- Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD (March 1994). Mutation of a mutL homolog in hereditary colon cancer. Science 263 (5153): 1625–9. Bibcode:1994Sci...263.1625P. PMID 8128251. doi:10.1126/science.8128251.
- Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, Igari T, Koike M, Chiba M, Mori T (November 1997). Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nature Genetics 17 (3): 271–2. PMID 9354786. doi:10.1038/ng1197-271.
- Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM (September 1994). Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 371 (6492): 75–80. Bibcode:1994Natur.371...75N. PMID 8072530. doi:10.1038/371075a0.
- Lu SL, Kawabata M, Imamura T, Akiyama Y, Nomizu T, Miyazono K, Yuasa Y (May 1998). HNPCC associated with germline mutation in the TGF-beta type II receptor gene. Nature Genetics 19 (1): 17–8. PMID 9590282. doi:10.1038/ng0598-17.
- Ou J, Rasmussen M, Westers H, Andersen SD, Jager PO, Kooi KA, Niessen RC, Eggen BJ, Nielsen FC, Kleibeuker JH, Sijmons RH, Rasmussen LJ, Hofstra RM (April 2009). Biochemical characterization of MLH3 missense mutations does not reveal an apparent role of MLH3 in Lynch syndrome. Genes, Chromosomes & Cancer 48 (4): 340–50. PMID 19156873. doi:10.1002/gcc.20644.
- Ramsoekh D, Wagner A, van Leerdam ME, Dinjens WN, Steyerberg EW, Halley DJ, Kuipers EJ, Dooijes D (November 2008). A high incidence of MSH6 mutations in Amsterdam criteria II-negative families tested in a diagnostic setting. Gut 57 (11): 1539–44. PMID 18625694. doi:10.1136/gut.2008.156695.
- Suchy J, Lubinski J (June 2008). MSH6 syndrome. Hereditary Cancer in Clinical Practice 6 (2): 103–4. PMC 2735474. PMID 19804606. doi:10.1186/1897-4287-6-2-103.
- Goldberg Y, Porat RM, Kedar I, Shochat C, Galinsky D, Hamburger T, Hubert A, Strul H, Kariiv R, Ben-Avi L, Savion M, Pikarsky E, Abeliovich D, Bercovich D, Lerer I, Peretz T (June 2010). An Ashkenazi founder mutation in the MSH6 gene leading to HNPCC. Familial Cancer 9 (2): 141–50. PMID 19851887. doi:10.1007/s10689-009-9298-9.
- http://www.genetics.edu.au/publications-and-resources/facts-sheets/fact-sheet-33-bowel-cancer-and-inherited-predisposition
- Giardiello, Francis M.; Allen, John I.; Axilbund, Jennifer E.; Boland, C. Richard; Burke, Carol A.; Burt, Randall W.; Church, James M.; Dominitz, Jason A. та ін. (August 2014). Guidelines on Genetic Evaluation and Management of Lynch Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 147 (2): 502–526. PMID 25043945. doi:10.1053/j.gastro.2014.04.001.
- Snowsill, Tristan; Huxley, Nicola; Hoyle, Martin; Jones-Hughes, Tracey; Coelho, Helen; Cooper, Chris; Frayling, Ian; Hyde, Chris (September 2014). A systematic review and economic evaluation of diagnostic strategies for Lynch syndrome. Health Technology Assessment 18 (58): 1–406. ISSN 1366-5278. PMC 4781313. PMID 25244061. doi:10.3310/hta18580.
- Snowsill, Tristan; Coelho, Helen; Huxley, Nicola; Jones-Hughes, Tracey; Briscoe, Simon; Frayling, Ian M.; Hyde, Chris (September 2017). Molecular testing for Lynch syndrome in people with colorectal cancer: systematic reviews and economic evaluation. Health Technology Assessment (Winchester, England) 21 (51): 1–238. ISSN 2046-4924. PMC 5611555. PMID 28895526. doi:10.3310/hta21510.
- Evrard, Camille; Tachon, Gaëlle; Randrian, Violaine; Karayan-Tapon, Lucie; Tougeron, David (15 жовтня 2019). Microsatellite Instability: Diagnosis, Heterogeneity, Discordance, and Clinical Impact in Colorectal Cancer. Cancers 11 (10): 1567. ISSN 2072-6694. PMC 6826728. PMID 31618962. doi:10.3390/cancers11101567.
- Hereditary Colorectal Cancer > Background. From Medscape. By Juan Carlos Munoz and Louis R Lambiase. Updated: Oct 31, 2011
- Burn J, Bishop DT, Mecklin JP, Macrae F, Möslein G, Olschwang S, Bisgaard ML, Ramesar R, Eccles D, Maher ER, Bertario L, Jarvinen HJ, Lindblom A, Evans DG, Lubinski J, Morrison PJ, Ho JW, Vasen HF, Side L, Thomas HJ, Scott RJ, Dunlop M, Barker G, Elliott F, Jass JR, Fodde R, Lynch HT, Mathers JC (December 2008). Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. The New England Journal of Medicine 359 (24): 2567–78. PMID 19073976. doi:10.1056/NEJMoa0801297.
- Aspirin Confers Long-Term Protective Effect in Lynch Syndrome Patients. Процитовано 7 листопада 2009.
- Vasen HF, Watson P, Mecklin JP, Lynch HT (June 1999). New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 116 (6): 1453–6. PMID 10348829. doi:10.1016/S0016-5085(99)70510-X.
- Vindigni, Stephen M.; Kaz, Andrew M. (2016). Universal Screening of Colorectal Cancers for Lynch Syndrome: Challenges and Opportunities. Digestive Diseases and Sciences 61 (4): 969–976. ISSN 0163-2116. PMID 26602911. doi:10.1007/s10620-015-3964-6.
- Bui, Quan M.; Lin, David; Ho, Wendy (February 2017). Approach to Lynch Syndrome for the Gastroenterologist. Digestive Diseases and Sciences 62 (2): 299–304. ISSN 0163-2116. PMID 27990589. doi:10.1007/s10620-016-4346-4.
- Boland CR, Koi M, Chang DK, Carethers JM (2007). The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside. Familial Cancer 7 (1): 41–52. PMC 2847875. PMID 17636426. doi:10.1007/s10689-007-9145-9.
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA (June 2015). PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. The New England Journal of Medicine 372 (26): 2509–20. PMC 4481136. PMID 26028255. doi:10.1056/NEJMoa1500596.
- Lynch HT, Shaw MW, Magnuson CW, Larsen AL, Krush AJ (February 1966). Hereditary factors in cancer. Study of two large midwestern kindreds. Archives of Internal Medicine 117 (2): 206–12. PMID 5901552. doi:10.1001/archinte.117.2.206.
- Bellizzi AM, Frankel WL (November 2009). Colorectal cancer due to deficiency in DNA mismatch repair function: a review. Advances in Anatomic Pathology 16 (6): 405–17. PMID 19851131. doi:10.1097/PAP.0b013e3181bb6bdc.
- Lindor NM (October 2009). Familial colorectal cancer type X: the other half of hereditary nonpolyposis colon cancer syndrome. Surgical Oncology Clinics of North America 18 (4): 637–45. PMC 3454516. PMID 19793571. doi:10.1016/j.soc.2009.07.003.
- Lindor NM, Rabe K, Petersen GM, Haile R, Casey G, Baron J, Gallinger S, Bapat B, Aronson M, Hopper J, Jass J, LeMarchand L, Grove J, Potter J, Newcomb P, Terdiman JP, Conrad P, Moslein G, Goldberg R, Ziogas A, Anton-Culver H, de Andrade M, Siegmund K, Thibodeau SN, Boardman LA, Seminara D (April 2005). Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 293 (16): 1979–85. PMC 2933042. PMID 15855431. doi:10.1001/jama.293.16.1979.
- Scott RJ, McPhillips M, Meldrum CJ, Fitzgerald PE, Adams K, Spigelman AD, du Sart D, Tucker K, Kirk J (January 2001). Hereditary nonpolyposis colorectal cancer in 95 families: differences and similarities between mutation-positive and mutation-negative kindreds. American Journal of Human Genetics 68 (1): 118–127. PMC 1234904. PMID 11112663. doi:10.1086/316942.
- Gologan A, Krasinskas A, Hunt J, Thull DL, Farkas L, Sepulveda AR (November 2005). Performance of the revised Bethesda guidelines for identification of colorectal carcinomas with a high level of microsatellite instability. Archives of Pathology & Laboratory Medicine 129 (11): 1390–7. PMID 16253017. doi:10.1043/1543-2165(2005)129[1390:POTRBG]2.0.CO;2.
- Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S (February 2004). Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. Journal of the National Cancer Institute 96 (4): 261–8. PMC 2933058. PMID 14970275. doi:10.1093/jnci/djh034.
- Lipton LR, Johnson V, Cummings C, Fisher S, Risby P, Eftekhar Sadat AT, Cranston T, Izatt L, Sasieni P, Hodgson SV, Thomas HJ, Tomlinson IP (December 2004). Refining the Amsterdam Criteria and Bethesda Guidelines: testing algorithms for the prediction of mismatch repair mutation status in the familial cancer clinic. Journal of Clinical Oncology 22 (24): 4934–43. PMID 15611508. doi:10.1200/JCO.2004.11.084. Архів оригіналу за 15 квітня 2013.
- Lynch Syndrome UK. Процитовано 31 березня 2018.
- Bowel Cancer UK: Lynch Syndrome. Процитовано 31 березня 2018.
- CDC: March 22nd is National Lynch Syndrome Awareness Day!. 20 березня 2018. Процитовано 31 березня 2018.