Хронічний лімфолейкоз
Хронічна лімфоцитарна лейкемія (ХЛЛ) або хронічний лімфоцитарний лейкоз — тип онкологічного захворювання крові, при якому червоний кістковий мозок утворює багато онкотрасформованих клітин лімфоїдної лінії — трансформованих проліферуючих попередників лімфоцитів (тип білих кров'яних клітин). Першопричиною ХЛЛ є мутація протоонкогену в клітинах лімфоїдної лінії червоного кісткового мозку з подальшою онкотрансформацією, утворенням злоякісного клону, що не знищується ніякими захисними механізмами: апоптозом, імунною системою. На початку патогенезу ХЛЛ симптомів зазвичай не буває. Пізніше можуть виникнути неболючі набряки лімфатичних вузлів, відчуття втоми, підвищення температури, нічне потовиділення або втрата ваги без ясних причин. Також може спостерігатися збільшення селезінки та низький рівень еритроцитів (анемія). Зазвичай стан пацієнта поступово погіршується роками. Фактори ризику включають спадковість — наявність серед предків випадків ХЛЛ. Цю патологію можуть викликати мутагени, зокрема різні пестициди, інсектициди. Патогенез ХЛЛ призводить до накопичення клітин лінії В-лімфоцитів у червоному кістковому мозку, лімфатичних вузлах та периферійній крові. Ці клітини не функціональні і витісняють здорові нормальні клітини крові. ХЛЛ ділиться на два основних типи з наступними двома типами злоякісних клітин:
- Клітини з мутантним геном IGHV.
- Клітини, шо мають мутації інших протоонкогенів.
| Хронічний лімфолейкоз | |
|---|---|
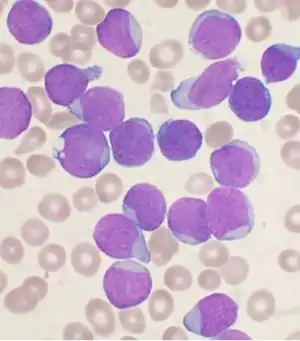 Мазок рідини з кісткового мозку пацієнта пофарбованою за Райтом з B-клітиною-попередницею гострої лімфобластної лейкемії Мазок рідини з кісткового мозку пацієнта пофарбованою за Райтом з B-клітиною-попередницею гострої лімфобластної лейкемії | |
| Спеціальність | Гематологія |
| Препарати | uracil mustardd[1], Алемтузумаб[1][2], меркаптопурин[1], Циклофосфамід[1][2], Хлорамбуцил[1][2], Пегаспаргаза[1], Кладрибін[1], Іфосфамід[1], etoposide phosphated[1], преднізон[2], Бендамустин[2], (RS)-lenalidomided[2], pentostatind[2], нілотиніб[2], Пегфілграстим[2], Доксорубіцин[2], dasatinib monohydrated[2], Ібрутиніб[2], idelalisibd[2], дексаметазон[2], venetoclaxd[2], Вінкристин[2], ритуксимаб[2], Офатумумаб[2], Обінутузумаб[2], Хлорамбуцил[3], Циклофосфамід[4] і venetoclaxd[5] |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | XH15T2 |
| МКХ-10 | C91.1 |
| МКХ-О | 9823 |
| OMIM | 109543 |
| DiseasesDB | 2641 |
| MedlinePlus | 000532 |
| eMedicine | /199313 |
| MeSH | D015451 |
| | |
Діагностика базується на аналізах крові, що дозволяють виявити велику кількість лімфоцитів на різних стадіях дозрівання в периферійній крові хворого. На ранніх стадіях патогенезу ХЛЛ перебіг захворювання безсимптомний, практикується лише спостереження за станом пацієнта, відомі методи втручання в перебіг захворювання можуть лише погіршити стан пацієнта і спровокувати негативні наслідки. Немає жодних фактів і випадків, коли втручання в перебіг захворювання на ранніх стадіях ХЛЛ мало б позитивні результати. Імунні дефекти виникають на початку перебігу ХЛЛ, збільшують ризик інфекцій, які слід належним чином лікувати антибіотиками. У пацієнтів з погіршення симптоматики та загостренням перебігу захворювання, перетворенням хронічної форми в гостру може застосовуватися хіміо- або імунотерапія. Станом на 2019 рік ібрутиніб є первинним рекомендованим препаратом. При загостренні перебігу ХЛЛ застосовуються препарати флударабін, циклофосфамід та ритуксимаб. У 2015 році на ХЛЛ захворіло близько 904 000 людей у всьому світі і власлідок ХЛЛ більше 60 700 з них померло. Захворювання найчастіше зустрічається у людей старших 50 років. Чоловіки хворіють на ХЛЛ вдвічі частіше за жінок. (співвідношення 6,8 : 3,5). ХЛЛ рідше трапляється в корінних жителів Азії, аніж в представників інших народів. Серед населення США через 5 років після встановлення діагнозу виживає 83 % пацієнтів. Серед загальної смертності від онкологічних захворювань смертність від ХЛЛ становить 1 %.
Ознаки та симптоми
Діагноз ХЛЛ ставиться на онові рутинного аналізу крові, що показує високий рівень лейкоцитів, високий вміст лімфоцитів серед лейкоцитів. На ранніх етапах перебігу ХЛЛ симптомів хвороби не спостерігається. Інколи на ранніх етапах патогенезу ХЛЛ спостерігаються збільшені лімфовузли з інвазією в лімфовузли патологічних лімфобластів. У таких випадках цю патологію називають ще лімфоцитарною лімфомою. В окремих випадках ХЛЛ ракові клітини заповнюють червоний кістковий мозок, що призводить до зниження рівня еритроцитів, нейтрофілів, тромбоцитів, що проявляється в анемії, імунодефіциті, порушенні згортання крові. На більш пізніх стадіях патогенезу ХЛЛ простежується лихоманка, нічні потовиділення, втрата ваги, підвищена втомлюваність. ХЛЛ можна розглядати як форму лімфоцитарної лімфоми або форму гострого лімфобластного лейкозу (ГЛЛ) з іншими клінічними проявами та іншим перебігом захворювання. При цьому при ХЛЛ ракові клітини виникають і поширюються з червоного кісткового мозку, а при лімфоцитарних лімфомах (ЛЛ) ракові клітини виникають і поширюються з лімфатичних вузлів. Вважається, що у всіх випадках ХЛЛ має моноклональний характер і його можна вважати формою моноклонального В-клітинного ліфмфобластного лейкозу (МВЛЛ). Цю форму лейкозу ще називають хронічним лімфобластн6им лейкозом типу МВЛЛ. Ця форма простежується як безсимптомна, хронічна зі збільшенням кількості В-клітинних лімфоцитів. Ці В-лімфоцити є аномальними: клон утворюється з однієї В-клітини, і всі клітини клону мають одні і ті ж білкові та імунологічні маркери, одні і ті ж хромосомні мутації та генні мутації.
Ускладнення
Ускладнення перебігу ХЛЛ можуть включати низький рівень антитіл крові (гіпогаммаглубулінемія), що призводить до імунодефіциту, рецидивів інфекційних захворювань, аутоімунної гемолітичної анемії (в 10 -15 % пацієнтів з ХЛЛ) та недостатності червоного кісткового мозку. ХЛЛ може трансформуватися в синдром Ріхтера, в прогресуючу дифузну великоклітинну В-лімфому, пролімфоцитарний лейкоз, лімфому Ходжкіна або в гострий лімфобластний лейкоз (ГЛЛ). Частота передтворення ХЛЛ в ГЛЛ становить близько 5 % випадків ХЛЛ. Можливе враження шлунково-кишківникового тракту, бактеріальні враження кишківника. Подібні ускладнення простежують часто при трансформації ХЛЛ в синдром Ріхтера. Але бувають випадки враження кишківника і без трансформації в синдром Ріхтера.
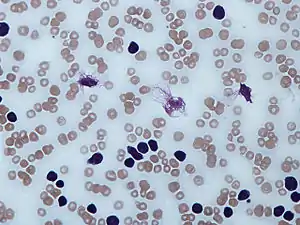
Діагностика
Підозра на діагнох ХЛЛ виникає внаслідок виявлення збільшення кількості лейкоцитів і вмісту лімфоцитів серед лейкоцитів в периферійній крові під час рутинного аналізу крові. Наявність високого лімфоцитозу, особливо в літніх людей, особливо якщо рівень лімфоцитів зростає в кілька разів, викликає підозру на наявність ХЛЛ. Тоді необхідно проводити додаткові діагностичні тести: проточну цитофлуориметрію, додаткові цитологічні дослідження, імінологічні дослідження, цитогенетичні дослідження. Якщо на препараті крові спостерігається велика кількість зруйнованих клітин – так званих «розмитих клітин», то це може свідчити про діагноз ХЛЛ. Такі «розмиті клітини» - це клітини з аномальним цитоскелетом, в якому бракує білка віментину. При ХЛЛ ракові клітини мають на поверхні маркери клітинної диференціації CD 5 та CD 23. Всі ракові клітини при ХЛЛ є одним клоном клітин – є генетично ідентичними. Ці аномальні попередники В-лімфоцитів різних стадій дозрівання не продукують нормальні антитіла. Нормальна популяція В-лімфоцитів поліклональна і продукує різні антитіла. Встановлення факту моноклональності популяції В-лімфоцитів периферійної крові є підставою для діагнозу ХЛЛ чи будь-якої іншої форми В-клітинного онкологічного захворювання, включно з В-клітинною неходжкінською лімфомою (НХЛ). Для встановлення діагнозу ХЛЛ необхідна комбінація мікроскопічного дослідження периферичної крові та аналізу лімфоцитів за допомогою проточної цитометрії для підтвердження клональності та експресії молекули маркера. І те, і інше легко досягти за допомогою невеликої кількості крові. Прилад проточного цитометра може досліджувати експресію молекул на окремих клітинах у рідинах. Це вимагає використання специфічних антитіл для маркування молекул флуоресцентними мітками, які розпізнає прилад. При ХЛЛ лімфоцити генетично клональні, з лінії В-клітин (експресують маркери молекул кластерів диференціації CD 19 і CD 20) і характерно експресують маркери молекул CD5 і CD23. Ці В-клітини нагадують нормальні лімфоцити під мікроскопом, хоч і трохи менші, і при розмазуванні на предметне скло крихкі, утворюючи безліч зруйнованих клітин, які називаються «розмитими клітинами». Імунологічні маркери ХЛЛ дозволяють ідентифікувати однорідну підгрупу класичної ХЛЛ, що відрізняється від атипової / змішаної ХЛЛ експресією п’яти маркерів (CD5, CD23, FMC7, CD22 та маркеру імуноглобулінового легкого ланцюга). Система оцінки ХЛЛ необхідна для диференціального діагнозу між класичною формою ХЛЛ та іншими В-клітинними хронічними лімфопроліферативними патологіями, але не для імунологічної різниці між змішаною / атиповою формами ХЛЛ та лімфомою мантійних клітин (злоякісні В-клітини MХЛ). Розрізнити ХЛЛ та МХЛ можна за допомогою маркерів CD54 та CD200. Серед рутинних маркерів найбільш виразною особливістю є коефіцієнт середньої інтенсивності флуоресценції CD20 / CD23.
Клінічні стадії
Стадія патогенезу, що визначає рівень захворювання, проводиться за допомогою системи Рея та Біне і базується, головним чином, на наявності низького рівня тромбоцитів та еритроцитів. Рання стадія захворювання не потребує лікування. ХЛЛ та ЛЛ вважаються одним і тим же основним захворюванням, лише з різними проявами.
Система стадій ХЛЛ Рея
- Стадія 0: характеризується високим лімфоцитозом (> 15 000 клітин на мм куб.) без лімфаденопатії, гепатоспленомегалії, анемії або тромбоцитопенії.
- I стадія: характеризується абсолютним лімфоцитозом з лімфаденопатією без гепатоспленомегалії, анемії або тромбоцитопенії
- II стадія: характеризується абсолютним лімфоцитозом з гепатомегалією або спленомегалією з лімфаденопатією або без неї.
- III стадія: характеризується абсолютним лімфоцитозом та анемією (гемоглобін < 11 г / дл) з лімфаденопатією або без неї, гепатомегалія або спленомегалія
- IV стадія: характеризується абсолютним лімфоцитозом і тромбоцитопенією (< 100 000 на мм куб.) з лімфаденопатією або без неї, гепатомегалією, спленомегалією або анемією.
Система стадій ХЛЛ Біне
- Клінічна стадія A: характеризується відсутністю анемії або тромбоцитопенії та менш ніж трьома зонами ураження лімфоїдів (стадії Рея 0, I та II)
- Клінічна стадія B: характеризується відсутністю анемії або тромбоцитопенії з трьома або більше ділянками ураження лімфоїдів (стадії Rai I і II)
- Клінічна стадія C: характеризується анемією та / або тромбоцитопенією незалежно від кількості ділянок збільшення лімфоїдної клітини (Rai III та IV стадії)
Каріотипування
Більшість випадків ХЛЛ супроводжуються хромосомними мутаціями, що зачіпають протоонкогени, локалізовані в тих чи інших хромосомах і є характерними для ХЛЛ: делеції del(13q14), del(11q23), del(17p) – ці мутації вважаються первинними і є поганим прогнозом перебігу захворювання. Низку інших хромосомних мутацій при ХЛЛ вважають вторинними. Мутація del(13q14) простежується у 15 – 20 % випадків ХЛЛ і зачіпає ген miR15a/16-1. Втрата цього гена порушує апоптоз клітин та регуляцію протоонкогену BCL-2.
Супутні захворювання
Раніше випадки захворювань крові з подібною цитологічною картиною, але з фенотипом Т-клітин, називали Т-клітинним ХЛЛ. Але зараз ці форми ХЛЛ визнані окремою групою захворювань і в даний час класифікуються як Т-клітинні пролімфоцитарні лейкози. ХЛЛ не слід плутати з гострим лімфобластним лейкозом (ГЛЛ) - вкрай агресивним лейкозом, який найчастіше діагностується у дітей і потребує інтенсивного лікування в стаціонарних умовах. Лімфоїдні розлади, які можуть проявлятися як хронічний лейкоз і які можна сплутати з типовим В-клітинним ХЛЛ:
- Фолікулярна лімфома
- Лімфома крайової зони селезінки
- Вузлова крайова зона В-клітинної лімфоми
- Мантійно-клітинна лімфома
- Пролімфоцитарний лейкоз (В-клітиний або Т-клітиний)
- Лімфоплазматична лімфома
- Синдром Сезарі
- Тліючий Т-клітинний лейкоз
- Лімфома у дорослих
- Гематологічні розлади, які можуть нагадувати ХЛЛ за своїм клінічним виглядом, поведінкою та мікроскопічним виглядом, включають лімфому мантійних клітин, лімфому крайової зони, пролімфоцитарний лейкоз В-клітин та лімфоплазматичну лімфому.
В-клітинний пролімфоцитарний лейкоз - більш агресивна патологія, наявні клітини з подібним фенотипом, але клітини значно більші за нормальні лімфоцити і мають помітне ядерце. Ця різниця важлива, оскільки прогноз та терапія відрізняються від ХЛЛ. Волохатоклітинний лейкоз - це також новоутворення В-лімфоцитів, але новоутворені клітини мають чітку морфологію під мікроскопом (клітини лейкозу волохатих клітин мають на своїх поверхнях ніжні, схожі на волосся виступи) та унікальну експресію маркерних молекул. Всі В-клітинні злоякісні пухлини крові та кісткового мозку можна диференціювати між собою поєднанням клітинної мікроскопічної морфології, експресії молекули маркера та специфічних пухлинно-асоційованих генних дефектів. Найкраще це досягається аналізом крові, кісткового мозку та клітин лімфатичних вузлів у пацієнта цитологом, що проходить спеціальну підготовку щодо патологій крові. Для аналізу клітинних маркерів необхідний проточний цитометр, і для виявлення генетичних проблем у клітинах може знадобитися візуалізація змін ДНК за допомогою флуоресцентних зондів за допомогою FISH-діагностики.
Лікування
Лікування ХЛЛ полягає в першу чергу у контролі над хворобою та її симптомами, а не на прямому лікуванні. Щодо тих пацієнтів, які не мають або мають лише мінімальні симптоми, проводиться лише ретельне спостереження. У випадку загострення перебігу хвороби ХЛЛ лікується хіміотерапією, променевою терапією, біологічною терапією або трансплантацією кісткового мозку. Іноді симптоми лікують хірургічним шляхом (спленектомія - видалення збільшеної селезінки) або променевою терапією (знищення ракових клітин в набряклих лімфатичних вузлах). Початкове лікування ХЛЛ залежить від точного діагнозу та прогресування захворювання, і навіть з урахуванням переваг та досвіду лікаря. ХЛЛ вважається невиліковним і прогресує повільно. Багато людей з ХЛЛ ведуть нормальне та активне життя протягом багатьох років - в деяких випадках десятиліть. Через повільний патогенез на ранніх стадіях ХЛЛ, переважно, не лікується, оскільки вважається, що раннє втручання ХЛЛ не покращує час виживання або якість життя. Натомість стан контролюється з часом, щоб виявити будь-які зміни в схемі захворювання. Рішення розпочати лікування ХЛЛ приймається, коли симптоми або показники крові у людини вказують на те, що хвороба прогресувала до такої міри, що вона може вплинути на якість життя. Клінічні стадії, такі як чотириступінчаста система Рея та класифікація стадій Біне, можуть допомогти визначити, коли і як лікувати пацієнта. Визначити, коли починати лікування та якими засобами часто важко, жодної переваги виживання не спостерігається при лікуванні захворювання на ранніх стадіях патогенезу. Робоча група Національного інституту раку США сформулювала настанови щодо лікування ХЛЛ з конкретними показниками, які слід виконувати до загострення захворювання.
Хіміотерапія
Комбіновані схеми хіміотерапії ефективні як при вперше діагностованому, так і при рецидиві ХЛЛ. Комбінації флударабіну з алкилирующими агентами (циклофосфамідом) дають вищі показники реакції та довшу виживання без прогресування, ніж окремі агенти:
- FC (флударабін з циклофосфамідом)
- FR (флударабін з ритуксимабом)
- FCR (флударабін, циклофосфамід та ритуксимаб)
- CHOP (циклофосфамід, доксорубіцин, вінкристин та преднізолон)
Хоча було показано, що пуриновий аналог флударабін забезпечує чудові показники відповіді на хлорамбуцил як первинну терапію, жодні дані не свідчать про те, що раннє використання флударабіну покращує загальну виживаність, і деякі клініцисти вважають за краще резервувати флударабін для рецидивів захворювання. Хемоімунотерапія FCR показала, що покращує рівень відповіді, виживання без прогресування та загальну виживаність у великому рандомізованому дослідженні у пацієнтів з ХЛЛ, відібраних для належної фізичної підготовленості. Це було перше клінічне випробування, яке показало, що вибір терапії першої лінії може покращити загальну виживаність пацієнтів з ХЛЛ. Алкілуючі агенти, дозволені для ХЛЛ, включають бендамустин і циклофосфамід.
Цільова терапія
Цільова терапія атакує ракові клітини як конкретну мішень, з метою не нашкодити нормальним клітинам. Цілеспрямовані препарати, що застосовуються при ХЛЛ, включають венетоклакс (інгібітор Bcl-2), ібрутиніб (інгібітор тирозинкінази Брутона), іделалізиб та дувелісіб (інгібітори деяких форм ферменту фосфоїнозитид 3-кінази), а також моноклональні антитіла проти CD20 (ритуксимаб), офатумумаб та обінутузумаб) та CD52 (алемтузумаб).
Трансплантація стовбурових клітин
Аутологічна трансплантація стовбурових клітин з використанням власних клітин реципієнта не застосовується. Щодо хворих на ХЛЛ молодого віку з великим ризиком смертності іноді розглядаються варіанти алогенної трансплантації кровотворних стовбурових клітин (HSCT) зі знищенням власних клітин червоного кісткового мозку хворого, але такі процедури мають високий ризик смертності.
Стійка до хіміотерапії форма ХЛЛ
Стійка до хіміотерапії форма ХЛЛ - це форма цього захворювання, при якій злоякісні лімфобласти не реагують на хіміотерапію, проявляють таку ж стійскість до хіміотерапії як і нормальні клітини. У цьому випадку розглядаються більш агресивні методи лікування, включаючи трансплантацію червоного кісткового мозку після застосування леналідоміду, флавопіридолу. Іноді застосовують моноклональне антитіло алемтузумаб (спрямоване проти CD52).
ХЛЛ та вагітність
Під час вагітності інколи виникають випадки на ХЛЛ. Лікування ХЛЛ в цьому випадку відкладають до закінчення вагітності, застосування терапії ХЛЛ під час вагітності може привести з високою імовірністю до вад розвитку плоду або до загибелі плоду чи викидня.
Прогноз
Прогностичними факторами перебігу ХЛЛ є типи генетичних мутацій, виявлених в ракових лімфобластах. Мутації гена IGHV є позитивними прогностичними факторами – середній час життя пацієнтів становить 20 - 25 років, тоді як інші мутації є гіршими прогностичними факторами: делеція del(13q14) знижує середню виживаність до 17 років, трисомія 12 хромосоми знижує середню виживаність до 9 – 11 років, такі самі показники зниження виживаності стосуються делеції del(11q23). Хоча прогноз перебігу ХЛЛ дуже мінливий і залежить від різних факторів, включаючи різні мутації, середня 5-річна відносна виживаність становить 86,1%. Довжина теломер вважається важливим прогностичним показником виживання.
Джерела
- O'Brien S., Gribben J. G. (2008). Chronic Lymphocytic Leukemia // CRC Press. p. 19. ISBN 9781420068962.
- Chronic Lymphocytic Leukemia Treatment // National Cancer Institute. 26 October 2017. Retrieved 19 December 2017.
- Chronic Lymphocytic Leukemia — Cancer Stat Facts // seer.cancer.gov. Retrieved 20 December 2017.
- Kipps T. J., Stevenson F. K. , Wu C. J., Croce C. M., Packham G., Wierda W. G., et al. (January 2017). Chronic lymphocytic leukaemia // Nature Reviews. Disease Primers. 3: 16096. doi:10.1038/nrdp.2016.96. PMC 5336551. PMID 28102226.
- Ferri F. F. (2017). Ferri's Clinical Advisor 2018 E-Book: 5 Books in 1. Elsevier Health Sciences. p. 750. ISBN 9780323529570.
- Vos Theo, Allen Christine, Arora Megha, Barber Ryan M., et al. (October 2016). Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990—2015: a systematic analysis for the Global Burden of Disease Study 2015 // Lancet. — 388 (10053): 1545—1602. doi:10.1016/S0140-6736(16)31678-6. PMC 5055577. PMID 27733282.
- Wang Haidong, Naghavi Mohsen, Allen Christine et al. (October 2016). Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980—2015: a systematic analysis for the Global Burden of Disease Study 2015 // Lancet. 388 (10053): 1459—1544. doi:10.1016/s0140-6736(16)31012-1. PMC 5388903. PMID 27733281.
- Boelens J., Lust S., Vanhoecke B., Offner F. (February 2009). Chronic lymphocytic leukaemia // Anticancer Research. 29 (2): 605–15. PMID 19331210.
- Hallek M., Shanafelt T. D., Eichhorst B. (April 2018). Chronic lymphocytic leukaemia. Lancet. 391 (10129): 1524–1537. doi:10.1016/S0140-6736(18)30422-7. PMID 29477250. S2CID 3517733.
- Stilgenbauer Stephan, Furman Richard R., Zent Clive S. (2015-05-01). Management of Chronic Lymphocytic Leukemia // American Society of Clinical Oncology Educational Book. - (35): 164–175. doi:10.14694/EdBook_AM.2015.35.164. ISSN 1548-8748. PMID 25993154.
- Chronic Lymphocytic Leukemia Treatment // National Cancer Institute. - 1 January 1980. Retrieved 19 February 2019.
- Hallek M. (September 2017). Chronic lymphocytic leukemia: 2017 update on diagnosis, risk stratification, and treatment // American Journal of Hematology. - 92 (9): 946–965. doi:10.1002/ajh.24826. PMID 28782884.
- NDF-RT
- Drug Indications Extracted from FAERS — doi:10.5281/ZENODO.1435999
- Inxight: Drugs Database
- Inxight: Drugs Database
- Inxight: Drugs Database