Клітинне старіння
Клітинне старіння (англ. cellular senescence) — перманентна зупинка клітинного циклу, під час якої клітина зберігає метаболічну активність. Може виникнути із ряду причин, таких як вкорочення теломер (реплікативне старіння) або інших типів ушкодження ДНК, надактивації онкогенів (старіння індуковане онкогенами, OIS) або втрати активності генів супресорів пухлин та інших стресових умов. Клітинне старіння, перш за все, є протипухлинним шляхом, що запобігає розмноженню клітин, які стали на шлях злоякісного переродження, проте також існують дані про те, що воно задіяне у процесах відновлення тканин. З іншого боку клітинне старіння може мати і негативні наслідки, зокрема робити внесок у старіння цілого організму, і навіть, у деяких випадках, стимулювати розвиток злоякісних новоутворень.
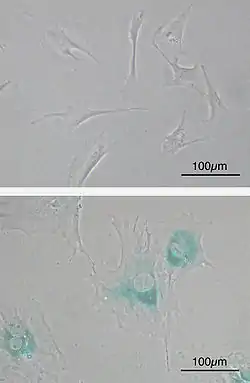
Історія дослідження
Вперше клітинне старіння було описане в 1965 році Гейфліком (англ. Hayflick) та Мургедом (англ. Moorhead). Вони помітили, що у культурі нормальні клітини втрачають здатність до проліферації через певну кількість поділів[1]. Кількість мітотичних поділів, які може здійснити клітина до завершення свого реплікативного життя, була названа межею Гейфліка[2]. 1990 року з'ясувалось, що причиною так званого реплікативного старіння є вкорочення теломер. Пізніше стали відомими інші фактори, що можуть викликати клітинне старіння, так у 1997 році Серрано із співробітниками описали старіння зумовлене надмірною мітогенною активацією через надекспресію RAS. Через 9 років було встановлено, що і в цьому випадку старіння виникає внаслідок ушкодження ДНК під час її інтенсивної реплікації. 2005 року опубліковано ряд статей, які доводять роль клітинного старіння як протипухлинного механізму, постарілі клітини виявлені у ряді людських неоплазій. Тоді ж з'явились відомості про те, що клітинне старіння може виникати не тільки внаслідок надактивації онкогенів, а й внаслідок втрати генів супресорів пухлин, зокрема PTEN. Подальші дослідження дозволили встановити, що процес старіння може потенційно використовуватись для терапії раку, оскільки він є важливим компонентом регресії пухлин після інактивації MYC, і постарілі клітини можуть бути мішенями для імунної системи[3].
Фактори, що спричинюють клітинне старіння
В дорослому організми багатоклітинних тварин наявні як мітотичні, так і пост-мітотичні клітини. Перші здатні до поділу, хоч більшість із них і перебувають у стані спокою (фаза G0), тобто тимчасово виходять із клітинного циклу, але можуть повертатись до проліферації у відповідь на дію певних стимулів. Пост-мітотичні клітини повністю втрачають здатність до поділу внаслідок диференціації, до них належать нейрони, м'язові клітини, еритроцити. Явище клітинного старіння стосується тільки мітотичних клітин, оскільки тільки вони можуть давати початок раковим пухлинам. Як постарілі, так і пост-мітотичні клітини незворотно втрачають здатність до проліферації, і хоч причини цього різні, проте деякі спільні ефектори можуть блокувати входження до клітинного циклу в обидвох випадках[4].
Вкорочення теломер
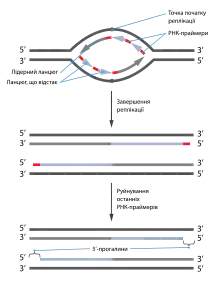
Першим із виявлених факторів, що можуть викликати клітинне старіння, є вкорочення теломер. Такий тип старіння називається реплікативним. Полімерази, що копіюють ДНК, здійснюють синтез тільки в одному напрямку: 5'→ 3', і приєднують нові нуклеотиди тільки до 3'-кінця вже наявних полінуклеотидів. Тому для роботи вони потребують РНК-праймерів, які розщеплюються по завершенню реплікації і замінюються на ДНК. Проте, коли ланцюг, що відстає, у реплікативній виделці досягає кінця ДНК, останній праймер руйнується, але не може бути замінений на ДНК через брак вільного 3'-кінця, що міг би бути використаний як субстрат для ДНК-полімерази, таким чином із кожним циклом реплікації теломерні (кінцеві) ділянки лінійних ДНК стають коротшими на 50—200 нуклеотидів[5][4]. Цього не спостергається у клітинах, що експресують теломеразу — зворотну транскриптазу, яка може відновлювати теломери. Види відрізняються між собою за кількістю клітин, в яких наявна теломераза. Так у мишей цей фермент активний у більшості клітин дорослого організму, тоді як у людей, він працює тільки в ембріональних стовбурових клітинах, деяких стовбурових клітинах зрілого організму, незначній кількості звичайних соматичних клітин, наприклад в активованих Т-лімфоцитах, а також багатьох ракових клітинах[2].
Функціональні теломери мають білкові кепи, що перешкоджають репараційним системам розпізнавати кінці хромосом як дволанцюгові розриви ДНК. Спроби репарувати кінці теломер могли би призвести до значної генетичної нестабільності через цикли злиття та розривів хромосом. Нефункціональні теломери все ж пригнічують такі спроби, проте запускають відповідь на ушкодження ДНК (англ. DNA damage response, DDR), ключовим компонентом якої є білок супресор пухлин p53. Оскільки вкорочені теломери в клітині не репаруються, це призводить до перманентної активації DDR, внаслідок чого наступає незворотна зупинка кілтинного циклу, тобто клітинне старіння[2]. Для його запуску достатньо тільки однієї нефункціональної теломери[4]
Старіння, індуковане вкороченням хромосом, інколи позначають як фаза M1 (англ. mortality phase 1). Клітини, які можуть ділитись у культурі необмежену кількість разів, називають реплікативно безсмертними. Хоча терміни «імморталізація» і «трансформація» історично вживались як синоніми, клітини можуть ставати безсмертними внаслідок індукції експресії теломерази, при цьому не набуваючи ознак злоякісної трансформації. Людські клітини можуть уникати реплікативного старіння внаслідок втрати функції p53, в такому разі вони розмножуються аж поки не досягнуть стану кризи або мітотичної катастрофи (M2, mortality phase 2), що характеризується вираженою генетичною нестабільністю і завершується смертю клітини[4].
Ушкодження ДНК
Вкорочення теломер — тільки один із видів порушення нормального функціонування геному, що може індукувати клітинне старіння. Такий же ефект можуть мати й ушкодження ДНК в інших місцях хромосом, зокрема потужними активаторами старіння є двониткові розриви ДНК, що виникають внаслідок дії іонізуючої радіації, інгібування топоізомерази та під дією інших факторів. Багато із цитотоксичних сполук, що використовуються для хемотерапії онкогенних захворювань, є агентами ушкодження ДНК і можуть викликати клітинне старіння як ракових, так і довколишніх клітин[2].
Оксидативний стрес може призводити до інших типів ушкодження ДНК, таких як модифікація основ або/і однониткові розриви. Проте під час реплікації або невдалих спроб репарації ці ураження також можуть перетворюватись у двониткові розриви[2]. Наприклад клітини мишей, попри те, що мають довгі теломери (>20 т.п.н.) і зазвичай експресують теломеразу, зазвичай старіють всього за кілька поділів при культивуванні у стандартних умовах. Це пояснюється концентрацією кисню (20%) вищою за фізіологічну. На відміну від людських клітин, мишачі дуже чутливі до таких умов, і високий вміст кисню призводить у них до розривів ДНК[4]. Існують дані про те, що оксидативний стрес може пришвидшувати вкорочення теломер, оскільки їхні G-багаті послідовності особливо чутливі до такого впливу[2].
Отже пряма чи опосередкована генерація двониткових розривів ДНК призводить до клітинного старіння, при чому встановлено, що навіть один такий розрив може викликати незворотну зупинку клітинного циклу, якщо не буде репарованим[2]. Розриви, як і втрата функціональних теломер, призводять до довготривалої активації DDR, внаслідок чого формуються γ-H2AX-позитивні осередки ушкодженої ДНК, пов'язані із старінням (англ. senescence-associated DNA damage foci, SDF), а також відбувається активація кіназ ATM і ATR, що через CHEK1 і CHEK1 передають сигнал на p53. У більшості клітин дещо пізніше вмикається також і сигнальний шлях p16INK4a, який регулює активність Rb[3][4].
Епігеномне ушкодження
Причиною виникнення клітинного старіння може стати епігенетичне ушкодження, зокрема масове перегрупування хроматину. Наприклад інгібітори гістондеацетилаз, що викликають перетворення ділянок гетерохроматину в еухроматин, є індукторами старіння. Таку ж реакцію може викликати знижена активність p300 гістонацетилтрансферази і c-Myc. В залежності від типу клітин порушення структури хроматину призводить або до запуску DDR попри брак справжніх ушкоджень ДНК, сигнал від якого йде на p53 і p21, або шляху p16INK4a[2][4].
Надто сильна або/і незбалансована стимуляція проліферації
Як механізм, що пригнічує злоякісне переродження, клітинне старіння може індукуватись у випадках, коли клітина отримує надто сильні, хронічні або/і незбалансовані мітогенні сигнали.
Старіння індуковане онкогенами
Старіння індуковане онкогенами або OIS вперше спостерігали in vitro при надекспресії онкогенної форми HRAS (цитоплазматичного білка, що бере участь у передачі мітогенних сигналів) — HRASG12V — у фібробластах людини. В таких умовах відбувається гіперрепілкація ДНК, що у свою чергу спричинює DDR специфічну для S-фази. Така відповідь хоч і відрізняється від DDR, викликаної двонитковими розривами ДНК або вкороченням теломер, але має з ними спільні ефектори (p53, який у цьому випадку активується через ARF, і p16INK4a) і також призводить до формування SDF[3]. Схожий ефект мають онкогенні версії або надекспресія інших членів сигнального шляху RAS (RAF, MEK, MOS і BRAF), а також ядерних білків, що сприяють проліферації, таких як E2F-1. OIS не відбувається при культивуванні клітин у середовищі без сироватки, що свідчить про те, що саме надлишкові мітогенні сигнали стимулюють старіння[4].
Більшість деталей функціонування OIS вивчались in vitro, але кілька мишачих моделей доводять, що цей процес справді важливий для супресії пухлин in vivo[3]. На користь такого припущення свідчить також і той факт, що у людей доброякісні лунини складаються переважно із постарілих клітин, що експерсують онкогенний BRAF. Для утворення злоякісних пухлин необхідні додаткові мутації, наприклад у генах p53 і p16INK4a[4].
Втрата функції супресорів пухлин
Щоб виконувати роль бар'єру для утворення злоякісних новоутворень, старіння повинно запускатись не тільки у випадку надактивації онкогнеів, а й втрати активності генів супресорів пухлин. Прикладом такої функції є PICS (англ. PTEN loss-induced cellular senescence), що активується у коли PTEN нормально не працює. PICS відрізняється від OIS браком гіперреплікації ДНК та DDR, і може розвиватись навіть коли входження в S-фазу і реплікація ДНК заблоковані афідіколіном[3].
Як і при активації клітинного старіння іншими факторами у PICS центральну роль відіграють сигнальні шляхи p53/p21 і p16INK4a, у цьому випадку перший шлях запускається через гіперактивацію mTOR, а другий — через ETS2[3].
Ознаки клітинного старіння
Зупинка проліферації
Основною ознакою постарілих клітин є незворотна зупинка проліферації, що відбувається внаслідок підвищеної експресії білків, що блокують проходження через клітинний цикл. Зазвичай постарілі клітини мають вміст ДНК, що відповідає фазі G1. Проте деякі онкогени спричинюють старіння із вмістом ДНК, характерним для фази G2, а ракові клітини можуть старіти із кількістю ДНК, що відповідає G2 або S фазі[4].
Щоб оцінити здатність клітин до поділу зазвичай використовують такі методи як визначення вмісту маркерів проліферації Ki67 і PCNA, мічення 5-бромодеоксіуридином (BrdU) або 3H-тимідином[6] або тест на здатність до колонієутворення[7]. Проте жоден із цих методів не дає можливості відрізнити постарілі клітини від пост-мітотичних або клітин у стані спокою (G0)[4].
Резистентність до апоптозу
Апоптоз, як і клітинне старіння, є протипухлинним механізмом, проте на відміну від старіння він забезпечує швидке знищення ушкоджених клітин, а не тільки зупинку проліферації. Багато постарілих клітин нечутливі до дії принаймні деяких із проапоптичних факторів. Наприклад, фібробласти людини у стані старіння не гинуть шляхом апоптозу у відповідь на дію цераміду (хоча така реакція може виникати у постарілих епітеліальних клітин), оксидативний стрес та позбавлення факторів росту, але зв'язування Fas рецепторів смерті із їхніми лігандами може викликати в них апоптоз[4].
До кінця не з'ясовано як клітини роблять вибір, відповідати на стрес старінням чи апоптозом. По-перше, це залежить від конкретного типу клітин, наприклад фібробласти й епітеліоцити більш схильні до старіння, а лімфоцити — до апоптозу. По-друге, — від природи та інтесивності стресу[4].
Примітки
- Hayflick L. (1965). The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 37: 614—36. PMID 14315085.
- Campisi J. (2012). Aging, Cellular Senescence, and Cancer. Exp Cell Res 75: Epub ahead of print. PMID 23140366. doi:10.1146/annurev-physiol-030212-183653.
- Nardella C, Clohessy JG, Alimonti A, Pandolfi PP (2011). Pro-senescence therapy for cancer treatment. Nat Rev Cancer 11: 503—11. PMID 21701512. doi:10.1038/nrc3057.
- Campisi J, d'Adda di Fagagna F (2007). Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8: 729—40. PMID 17667954. doi:10.1038/nrm2233.
- Hardin J, Bertoni G, Kleinsmith LJ (2011). Becker’s world of the cell (вид. 8th). Benjamin Cummings. с. 564—565. ISBN 0-321-71602-7.
- Collado M, Serrano M (2010). Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10: 51—57. PMID 20029423. doi:10.1038/nrc2772.
- Mumcuoglu M, Bagislar S, Yuzugullu H et al (2010). The ability to generate senescent progeny as a mechanism underlying breast cancer cell heterogeneity. PLoS One 5: e11288. PMID 20585577. doi:10.1371/journal.pone.0011288.