Лімфома Беркітта
Лімфома Беркітта (англ. Burkitt's lymphoma) — рак лімфатичної системи, зокрема B-лімфоцитів, який розвивається в гермінативному центрі[3]. Загальний рівень вилікування лімфоми Беркітта в розвинених країнах становить близько 90 %, але гірший у країнах з низьким рівнем доходу. Лімфома Беркітта є рідкістю у дорослих, де вона має гірший прогноз.
| Лімфома Беркітта | |
|---|---|
 Мікропрепарат пухлини з наявністю злоякісних клітин, забарвлення по Райту | |
| Спеціальність | Гематологія |
| Причини | Інфекція, яку спричинює вірус Епштейна — Барр |
| Препарати | Циклофосфамід[1] і Циклофосфамід[2] |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | 2A85.6 |
| МКХ-10 | C83.7 |
| МКХ-О | M9687/3 |
| OMIM | 113970 |
| DiseasesDB | 1784 |
| MedlinePlus | 001308 |
| eMedicine | med/1447602 |
| MeSH | D002051 |
| | |
Етимологія
Названо на честь Денніса Парсонса Беркітта, ірландського хірурга, який вперше описав хворобу в 1958 році, працюючи в Екваторіальній Африці[4].
Класифікація
За клінічними варіантами
Наразі хвороба поділяється на 3 клінічні варіанти:
- ендемічний;
- спорадичний;
- асоційований з імунодефіцитом.
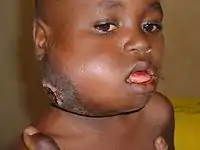
Ендемічний варіант (його ще називають «африканський варіант») найчастіше зустрічається у дітей, які живуть у ендемічних по малярії регіонах світу (наприклад, в Екваторіальній Африці, Бразилії та Папуа-Новій Гвінеї) між широтами 10 ° на південь та 10 ° на північ. Захворюваність у цих регіонах ендемічної хвороби становить 100 на мільйон дітей. Ураження вірусом Епштейна — Барр там зустрічається майже у всіх дітей. Вважається, що тривала малярія знижує стійкість до неї, дозволяючи їй злоякісно прогресувати. Лімфома уражає щелепи або інші лицьові кістки, дистальні відділи тонкої кишки, сліпої кишки, яєчники, яєчка, нирки або груди, центральну нервову систему.
Спорадичний варіант (також відомий як «неафриканський») є найпоширенішим варіантом, що зустрічається в регіонах, де малярія не є ендемічною. Пухлинні клітини мають схожий вигляд на ракові клітини ендемічного варіанту лімфоми Беркітта. Спорадичні лімфоми рідко асоціюються з вірусом Епштейна — Барр. Щелепа уражена рідко порівняно з ендемічним варіантом. Ілеоцекальна область є частою локалізацією процесу. Чоловіки уражаються в 2-3 рази частіше, ніж жінки (співвідношення чоловік-жінка, 2-3:1). Захворюваність на лімфому Беркітта у дорослих (> 20 років) була приблизно в 5 разів вища, ніж частота захворювання у дітей (<20 років).
Варіант, асоційований з імунодефіцитом, як правило розвивається при ВІЛ-інфекції або виникає при лікуванні пацієнтів після трансплантації, які приймають імуносупресивні препарати. Лімфома Беркітта ще може бути одним із захворювань, з яких може починатися СНІД. Близько 30-40 % випадків, пов'язаних з неходжкінськими лімфомами при ВІЛ-інфекції, спричинює лімфома Беркітта, і показник не зменшується з появою високоактивної антиретровірусної терапії ВІЛ-інфекції. Ця лімфома, як правило, уражає пацієнтів з високим рівнем CD4 (> 200 / мм3). Це свідчить про те, що зниження імунітету не є головним фактором ризику в цьому варіанті лімфоми Беркітта.
Система класифікації Національного інституту раку (США)
Включає:
- A — одинична пухлина за межами черевної порожнини;
- AR — внутрішньочеревна, більше 90 % пухлини резецировано;
- В — множинні пухлини за межами черевної порожнини;
- C — внутрішньочеревна пухлина;
- D — внутрішньочеревна пухлина + 1 або більше за межами черевної порожнини.
Система класифікації Енн-Арбор та Сент-Джуда / Мерфі
Включає 4 стадії:
- І стадія:
- Одинична пухлина (за межами лімфовузла);
- Одинарна анатомічна область (вузлова).
- Стадія II:
- Одинична пухлина із залученням регіонального вузла;
- Первинна пухлина шлунково-кишкового тракту;
- Лімфома, що включає вузлові ділянки з тієї ж сторони діафрагми.
- Стадія IIR складається з повністю резекованого внутрішньочеревного утворення.
- Стадія III:
- Лімфома, що включає ділянки з протилежних сторін діафрагми;
- Всі первинні внутрішньогрудні пухлини;
- Всі параспінальні або епідуральні пухлини;
- Екстенсивна внутрішньочеревна пухлина.
- Стадія IV складається з будь-якого з вищезазначених з ураженням ЦНС або кісткового мозку (<25 %).
Пацієнти з низьким рівнем ризику мають пухлини <10 см, захворювання на ранній стадії (I або II), хорошу працездатність та нормальну активність лактатдегідрогенази (ЛДГ). До пацієнтів з високим рівнем ризику належать усі інші пацієнти.
Патогенез
Точна причина та патофізіологічні механізми, що призводять до розвитку лімфоми Беркітта, невідомі. Існує кілька теорій.
Відомо, що вона являє собою високоагресивну В-лімфоцитну негоджкінську лімфому, що характеризується транслокацією та дерегуляцією гена c-myc на хромосомі 8. Лімфоми Беркітта притаманний швидкий і агресивний клінічний перебіг з частим ураженням кісткового мозку та центральної нервової системи (ЦНС).
Роль вірусу Епштейна—Барр
Цей вірус є членом родини герпесвірусів, який сильно пов'язаний з ендемічним варіантом лімфоми Беркітта, тоді як лише близько 20 % випадків спорадичного варіанту асоціюються з вірусом Епштейна—Барр. Він спричинює інфікування В-лімфоцитів, частина з яких ухиляється від Т-клітинної імунної відповіді та потрапляє у зародковий центр. Згодом це призводить до надмірної проліферації В-клітин. Показано, що EBNA-1 (ядерний антиген вірусу Епштейна—Барр) та декодуючі РНК мають слабкі антиапоптотичні властивості. Крім того, EBNA-3A та EBNA-3C можуть інгібувати експресію антиапоптотичного білка BCL-2.
Роль малярії
Малярійна інфекція також, ймовірно, відіграє роль у патогенезі лімфоми Беркітта, оскільки може призвести до пригнічення імунної відповіді, специфічної для цієї хвороби. Однак точний механізм опосередкованого лімфомагенезу недостатньо вивчений, але існують докази значної взаємодії між вірусною та клітинною мікроРНК, що перешкоджає нормальній експресії та трансляції генів. Лімфому Беркітта можна виявити у 25-40 % випадків, пов'язаних з імунодефіцитом.
Генетичні особливості
Класична зворотна транслокація t (8; 14) (q24; q32) (85 % випадків) призводить до транспозиції прото-онкогену c-myc на хромосому 8 одним із генів важкого ланцюга імуноглобуліну на хромосомі 14, в результаті чого відбувається активація гена c-myc і вважається відповідальним за проліферацію пухлини. Транслокація t (8; 14) є найпоширенішою, присутньою у 80 % випадків лімфоми Беркітта. У всіх інших випадках c-myc був переміщений близько до одного з генів легкого ланцюга імуноглобуліну на хромосомі 2 (легкий каппа-ланцюг) (t 8; 2) або 22 (легкий лямбда-ланцюг) (t 8; 22). Перевиробництво c-myc може змінити лімфоцити в ракові клітини, але й інші мутації генів можуть бути відповідальними за прогресування лімфоми Беркітта. Показано, що порушення гена p53 та смерть-пов'язаної протеїнкінази (DAP-кінази) сприяють зменшенню апоптозу.
C-myc — фактор роздягаючої транскрипції лейцину, який впливає на різні шляхи, регулює клітинний цикл, ріст, адгезію, диференціювання та апоптоз. Він надмірно експресується через його поєднання з підсилювачами генів імуноглобуліну. Такі гени, як циклін D2, TRAP1 та HLA-DRB1, індукуються надмірною експресією c-myc, тоді як інші, такі як p21 та похідний від тромбоцитів рецептор PDGFRA, послідовно репресуються, можливо, граючи роль у патогенезі хвороби.
E2F1 є членом групи факторів транскрипції E2F, яка бере участь у регуляції росту клітин. Останніми роками в більшості спорадичних випадків лімфоми Беркітта було виявлено, що активність E2F1 є надмірною.
Мутації гена ID3 виявлено у 34-68 % хворих. Роль цих інактивуючих мутацій ID3 може полягати у значному посиленні дії транслокації c-myc–імуноглобуліну, збільшення прогресування клітинного циклу та клітинної проліферації в лімфомних клітинах Беркітта. У дослідженні, яке передбачало генетичне секвенування лімфоми Беркітта, ID3 був одним із 70 генів, які мутували, включаючи ще GNA13, RET, PIK3R1, ARID1A та SMARCA4.
Клінічні прояви
Обумовлені локалізацією і об'ємом пухлини. Найбільш часто уражаються органи черевної порожнини: тонка кишка (частіше її термінальний відділ), брижа, а також шлунок, товста кишка, очеревина, печінка, селезінка. Специфічне ураження кісткового мозку та центральної нервової системи спостерігається в чверті випадків. Типовим є залучення нирок, яєчників, яєчок, абдомінальних і заочеревинних лімфатичних вузлів (особливо в ілеоцекальній області), рідше периферичних лімфатичних вузлів. У 10-15 % випадків відзначаться залучення лімфоглоткового кільця Вальдейера, слинних залоз, верхньої та нижньої щелепи, орбіти. Середостіння уражається рідко. Спостерігається спленомегалія (збільшення селезінки). Часто швидко розвивається втрата маси тіла (до 10 кг і більше за місяць). Характерним симптомокомплексом є розвиток синдрому «гострого живота» як результат обструктивної кишкової непрохідності, гострого апендициту, шлунково-кишкової кровотечі або перфорації внаслідок специфічної інфільтрації шлунка і / або кишечнику. При клінічному обстеженні у хворого можна виявити збільшення живота за рахунок пухлини і асциту. Зростання пухлини відбувається стрімко, і в більшості випадків до моменту звертання до лікаря ураження органів черевної порожнини являє собою масивний пухлинний конгломерат, з залученням декількох внутрішніх органів. Як правило це знаменує собою тяжкій або вкрай тяжкій стан, обумовлений великою пухлинною масою, інтоксикацією, виснаженням аж до кахексії, електролітними порушеннями. Нерідко основною терапевтичною проблемою у хворих на першому етапі розвитку хороби є прогресуюча гостра ниркова недостатність. Найчастішими причинами її розвитку є специфічне ураження нирок, синдром гострого лізису пухлини і порушення виходу сечі за рахунок здавлення сечоводів пухлинним конгломератом з розвитком постренальной анурії.
Ускладнення
Ймовірність ускладнень зростає зі ступенем захворювання. Через надзвичайно швидкі темпи росту пухлинних клітин, масове гостре руйнування пухлинних клітин під час початкової хіміотерапії може сформуватися синдром лізису пухлини, що потребує ниркового діалізу.
Діагностика
Основним в діагностиці є біопсія пухлинного утворення. Пухлина морфологічно характеризується дифузним ростом мономорфних клітин середнього розміру з округлими ядрами, з вузьким обідком базофільною цитоплазми. Типова картина «зоряного неба» (макрофаги з уламками ядер в цитоплазмі). Пухлина має імунофенотип CD20 +, CD10 +, CD38 +, BCL-6 +, BCL-2, CD44-, TdT-, CD3-. Індекс проліферативної активності пухлинних клітин Ki-67 наближається до 100 %. Особливості, які виключають діагноз лімфоми Беркітта, включають реаранжировку гена BCL-6 (незалежно від ядерного фарбування BCL-6), позитивність BCL-2, наявність t (14; 18) та індекс Ki-67 менше 95 %.
Лікування
Головним є хімієтерапія. У лікуванні лімфоми Беркітта не мають значення оперативне втручання та променева терапія. Важливо уважно стежити за біохімічними показниками крові під час хімієтерапії через високий ризик синдрому лізису пухлини та нефропатії внаслідок дії сечової кислоти. Підтримувати адекватну гідратацію через внутрішньовенне введення рідини слід починаючи за 24 години до призначення хімієтерапії. Необхідно підтримати високий відтік сечі (200—250 мл / м2 / годину) і уважно стежити за функцією нирок. Більшість пацієнтів слід попередньо лікувати розбуриказою, яка вводиться в дозі 0,0002 г / кг внутрішньовенно щодня впродовж 5 днів. Пацієнти нижчого ризику та ті, хто не переносить розбуриказу, можуть приймати алопуринол по 0,3 г двічі на день.
У перші дні лікування слід слідкувати за кількістю еритроцитів у крові, факторами коагуляції та принаймні двічі на день вимірювати рівень сечової кислоти, калію, кальцію, фосфору, магнію та креатиніну в сироватці крові, контролювати активність АлАТ і АсАТ. Хворий має бути підключеним до кардіомонітору. Лікування повинно проводитися в медичному закладі, де доступний нирковий діаліз (гемодіаліз).
Більшість схем хімієтерапії були спочатку прийняті з педіатричних протоколів дослідження, в яких використовувались кілька відомих діючих агентів, включаючи циклофосфамід, вінкристин, високодозований метотрексат, доксорубіцин, етопозид та цитарабін. Інші протоколи включають модифікований фракціонований циклофосфамід, вінкристин, доксорубіцин та дексаметазон, застосовуються переважно у дорослих. Застосування цих схем призводить до виліковування щонайменше 60 % хворих.
Застосування внутрішньочеревного метотрексату з цитарабіном або без нього, та гідрокортизоном, включена до більшості схем лікування. Без їхнього введення у половини пацієнтів розвинеться рецидив ураження лімфомою мозку.
Однією з сучасних схем хімієтерапії є «CODOX-M / IVAC» (циклофосфамід, вінкристин, доксорубіцин, високодозований метотрексат / іфосфамід, етопозид, цитарабін у високій дозі) складається з 4 циклів, кожен цикл триває до відновлення абсолютної кількості нейтрофілів > 1000 клітин / мкл; тромбоцитів > 109 кл. / мкл. Цикли 1 і 3 включають CODOX-M, а цикли 2 і 4 включають IVAC. Трьох циклів CODOX-M зазвичай достатньо хворих з низьким рівнем ризику, тоді як пацієнти з високим рівнем ризику отримують 4 загальних циклу (2 цикли CODOX-M, що чергуються з 2 циклами IVAC).
Схема CODOX-M включає:
- Циклофосфамід 0,8 г / м2 внутрішньовенно (в/в) у 1-й день, далі 200 мг / м2 в/в у дні 2-5-й;
- Доксорубіцин 0,04 г / м2 в/в у 1-й день;
- Вінкристин 0,0015 г / м2 в/в (без обмеження дози) у 1 та 8-й дні (цикл 1), а також у дні 1, 8 та 15-й (цикл 3)
- Метотрексат 1,2 г / м2 в/в упродовж 1 години 10-го дня; потім 0,24 г / м2 / год упродовж наступних 23 годин; лейковорин вводиться через 36 годин від початку введення метотрексату;
- Інтратекально цитарабін 0,07 г (якщо пацієнт старше 3 років) на 1 та 3-й дні;
- Інтратекально метотрексат 0,0012 г (якщо пацієнт старше 3 років) на 15 день.
Схема IVAC включає:
- Іфосфамід 1,5 г / м2 в/в у 1-5-й дні;
- Етопозид 0,06 г / м2 в/в у 1-5-й дні;
- Цитарабін 2 г / м2 в/в кожні 12 годин у 1-2-й дні
- Інтратекальний метотрексат 0,0012 г (якщо пацієнт старше 3 років) на 5-й день.
Введення колонієстимулюючих факторів зазвичай починають через 24 години після закінчення хімієтерапії і тривають до тих пір, поки кількість нейтрофілів не досягне 1000 клітин в 1 мкл.
Якщо зафіксовано ураження центральної нервової системи, пацієнти отримують інтенсивніший інтратекальний режим протягом циклів 1 і 2. Цитарабін 0,07 г призначається в 1, 3 і 5-й дні цикл 1, а також на 7 та 9-й день циклу 2. Крім того, вводиться інтратекальний метотрексат 0,00125 г на 15 та 17-й день (цикл 1) та 5-й день (цикл 2). Для циклів 3 і 4 застосовуються звичайні профілактичні внутрішньочеревні дози цитозину арабінозиду та метотрексату.
Схема гіпер-CVAD (модифікований фракціонований циклофосфамід, вінкристин, доксорубіцин та дексаметазон) складається з чергування циклів A (1, 3, 5 та 7-й) та B (2, 4, 6 та 8-й). Другий та наступні цикли проводяться тоді, коли кількість лейкоцитів у пацієнта більше 3000 / мм3, а кількість тромбоцитів вище 60000 / мм3 (між 14 і 21 днями). Цикл А включає:
- Циклофосфамід 0,3 г / м2 в/в впродовж 2-х годин кожні 12 годин перші 6 доз, на 1-3-й дні;
- Месна 0,6 г / м2 шляхом безперервної інфузії впродовж 24 годин, починаючи за 1 годину до циклофосфаміду та продовжуючи впродовж 12 годин після останньої дози циклофосфаміду;
- Вінкристин 0,002 г / доза в/в упродовж 2–3-х хвилин упродовж 4 та 11-го днів;
- Доксорубіцин 0,05 г / м2 в/в упродовж 2-х годин 4-го дня;
- Дексаметазон 0,040 г / добу перорально або в/в 8 доз у 1-4-й дні та 11-14-й дні;
- Філграстим 10 мкг / кг / добу підшкірно, починаючи через 24 години після останньої дози хімієтерапії та продовжуючи до тих пір, поки кількість лейкоцитів не досягне 3000 кл / мм3.
Цикл B включає наступне:
- Метотрексат 1 г / м2 шляхом безперервної в/в інфузії впродовж 24 годин у 1-й день;
- Цитарабін 3 г / м2 в/в упродовж 2 годин кожні 12 годин 4 дози, на 2-3-й день;
- Кальцію лейковорин 0,05 г в/в упродовж 20 хвилин, що вводиться через 12 годин після завершення інфузії метотрексату, після чого через 6 годин лейковорин 0,015 г в/в кожні 6 годин 8 загальних доз або до тих пір, поки концентрація метотрексату в крові не буде <0,1 мкмоль / л;
- Філграстим 10 мкг / кг / добу, починаючи через 24 години після останньої дози хімієтерапії та продовжуючи, поки кількість лейкоцитів не досягне 3000 / мм3.
Дози лейковорину кальцію слід збільшувати до 0,05 г в/в кожні 6 годин, якщо концентрації метотрексату в сироватці крові такі:
- Більше 20 мкмоль / л наприкінці інфузії метотрексату (24 година);
- Більше 1 мкмоль / л через 24 години після закінчення інфузії метотрексату (48 година);
- Більше 0,1 мкмоль / л через 48 годин після закінчення інфузії метотрексату (72 година).
При ураженні центральної нервової системи метотрексат вводиться через люмбальну пункцію у спинномозковий канал на 2-й день кожного циклу по 0,0012 г, а цитарабін — в дозі 0,1 г кожного 7-го дня протягом 8 циклів.
Ритуксимаб — препарат, що містить рекомбінантні антитіла, які націлюють CD20 на поверхню В-лімфоцитів. Багато досліджень включають ритуксимаб у лікування лімфоми Беркітта.
Наразі проводяться дослідження інших схем, зокрема, CALGB 9251.
Після закінчення хімієтерапії слід оцінити відповідь на лікування: провести лабораторні дослідження (дослідження клінічного аналізу крові, ниркові та печінкові біохімічні показники, активність лактатдегідрогенази тощо), а також результати КТ.
Прогноз
Приблизно 90 % дітей і до 50-60 % дорослих, які хворіли на лімфому Беркітта та отримували поточні режими інтенсивної хімієтерапії, виживають тривало. Більшість рецидивів виникають упродовж першого року лікування лімфоми Беркітта. Недосягнення повної ремісії є дуже поганою прогностичною ознакою. Ті, хто звільнилися від проявів на 10-12 місяців, вважаються вилікуваними, хоча повідомлення про появу рецидивів після першого року лікування були описані серед африканської популяції та пацієнтів із одночасним ураженням вірусом імунодефіциту людини. Більшість пацієнтів цієї групи погано реагують на «терапію врятування», хоча деякі з них мають довгострокову виживаність. Режим цієї терапії, як правило, включає хімієтерапевтичні засоби, якими пацієнт раніше лікувався.
До появи сучасних агресивних терапевтичних програм діти з лімфомою Беркітта швидко помирали. При належному керуванні метаболічними наслідками швидкого обороту клітин та при комбінованій хімієтерапії та профілактиці ураження центральної нервової системи рівень виживання значно покращився до ≤ 60 %. Діти з обмеженою (A, AR; I та II стадією) хворобою мають чудовий прогноз із виживаністю більше 90 %. Пацієнти з більш обширною хворобою (III та IV стадії), особливо із залученням кісткового мозку та центральної нервової системи, мають гірший прогноз, але тривалість виживання в 50-90 % може бути досягнута при більш агресивних режимах хімієтерапії. Пацієнти з рецидивом мають тривалість виживання 20-50 %. Для тих, у кого спостерігається рецидив, у 25 % дітей можливо досягти тривалого виживання шляхом аутологічної трансплантації високих доз гематопоетичних стовбурових клітин. Додавання ритуксимабу до цієї терапевтичної схеми може додатково збільшити швидкість відповіді.
Дорослі з лімфомою Беркітта, особливо ті, хто має стадію запущеної стадії, хворіють гірше, ніж діти. Крім того залучення кісткового мозку зі складною цитогенетикою має гірший прогноз у ретроспективному спостереженні, що є більш поширеним серед пацієнтів літнього віку.
Система прогностичного оцінювання, розроблена в 2013 році, допомагає кількісно оцінити потенціал вилікування у недавно діагностованих дорослих пацієнтів з лімфомою Беркітта. Бали присвоюються наступним чином:
- Вік 40-59 років або приналежність до чорної раси будь-якого віку: 1 бал;
- Вік 60-79 років або III / IV стадія захворювання: 2 бали;
- Вік 80 років і старше: 4 бали.
Виділяють 4 групи ризику, засновані на системі балів:
- Низький ризик (5-річна відносна виживаність: 71 %): 0-1 бали;
- Низький проміжний (5-річна відносна виживаність: 55 %): 2 бали;
- Високий проміжний (5-річна відносна виживаність: 41 %): 3 бали;
- Високий ризик (5-річна відносна виживаність: 29 %): 4 і більше балів.
Примітки
- NDF-RT
- Inxight: Drugs Database
- Це зона лімфоїдного вузлика (фолікула) периферичної лімфоїдної тканини, в якій зрілі В-лімфоцити, активовані антигеном, проліферують, диференціюються і зазнають процеси соматичного гіпермутагенезу
- Burkitt D (1958). «A sarcoma involving the jaws in African children». The British Journal of Surgery. 46 (197): 218–23. doi:10.1002/bjs.18004619704. PMID 13628987 (англ.)
Джерела
- Ali H Kanbar, Ronald A Sacher Burkitt Lymphoma and Burkitt-like Lymphoma Updated: Apr 08, 2016 Medscape. Drugs & Diseases. Hematology (Chief Editor: Emmanuel C Besa) (англ.)
- Molyneux E, Rochford R, Griffin B, Newton R, Jackson G, Menon G, Harrison C, Israels T, Bailey S (April 2012). «Burkitt's lymphoma». The Lancet. 379 (9822): 1234—1244. doi:10.1016/S0140-6736(11)61177-X. PMID 22333947 (англ.)
- Whonamedit?- A dictionary of medical eponyms. Burkitt's lymphoma (англ.)
