Хвороба Вільсона
Хворо́ба Ві́льсона (англ. Wilson's disease; синоніми: гепато-лентикулярна дегенерація, гепатоцеребральна дистрофія, псевдосклероз Вестфаля) — спадкове захворювання з аутосомно-рецесивним типом успадкування![]() , при якому мідь накопичується у тканинах. Проявляється у вигляді неврологічних та/або психіатричних
, при якому мідь накопичується у тканинах. Проявляється у вигляді неврологічних та/або психіатричних![]() симптомів й уражень печінки
симптомів й уражень печінки![]() [4]. Хворобу Вільсона лікують медикаментами
[4]. Хворобу Вільсона лікують медикаментами![]() , що зменшують абсорбцію міді або видаляють її надлишок з організму. У деяких випадках необхідна трансплантація
, що зменшують абсорбцію міді або видаляють її надлишок з організму. У деяких випадках необхідна трансплантація![]() печінки.
печінки.
| Хвороба Вільсона | |
|---|---|
 Кільце Кейзера — Флайшера у пацієнта з хворобою Вільсона Кільце Кейзера — Флайшера у пацієнта з хворобою Вільсона | |
| Спеціальність | ендокринологія |
| Симптоми | набряк, жовтяниця, зміна особистостіd, blue nailsd і Обличчя великої панди |
| Причини | генетика |
| Препарати | Триетилентетрамін[1][2], D-penicillamined[2] і Триетилентетрамін[3] |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | 5C64.00 |
| МКХ-10 | E83.0 |
| OMIM | 277900 |
| DiseasesDB | 14152 |
| MedlinePlus | 000785 |
| eMedicine | neuro/570 |
| MeSH | D006527 |
| | |
Захворювання виникає через мутації у гені протеїну ATP7B, АТФ-ази P-типу, що транспортує катіони міді[4]. Одна ненормальна копія цього гену наявна в 1 зі 100 людей, у яких не розвинулись симптоми хвороби, — так званих носіїв хвороби. У дитини хвороба Вільсона може розвинутись лише у тому випадку, коли вона успадкує ген хвороби від обох батьків. Зазвичай симптоми починають проявлятися у віці від 6 до 20 років, але описані випадки у набагато старших людей. Хворобу Вільсона виявляють в 1-4 зі 100 000 людей[5]. Її названо на честь Самуеля Александра Кіннієра Вільсона![]() .
.
Історія

Захворювання названо на честь британського лікаря Самюеля Александра Кіннієра Вільсона (1878—1937), невролога, який 1912 року описав цей стан та патологічні зміни, що відбуваються при ньому в мозку та печінці[6]. Роботі Вільсона передували відомості від німецького невролога Карла Вестфаля (у 1883 році), який назвав цей стан «псевдосклерозом», британського невролога Вільяма Ґоверса (1888 року) та Адольфа Штрюмпеля (1898 року), який зауважив, що при такому стані розвивається цироз печінки[7]. Невропатолог Джон Натаніель Кернінґс 1948 року пов'язав накопичення міді у печінці з таким накопиченням у мозку[8]. Прояв гемолізу було описано 1968 року[9].
Кернінґс, а одночасно з ним і новозеландський невролог Дерек Денні-Браун, працюючи у Сполучених Штатах, 1951 року першим заявив про успішне лікування хвороби хелатором металів димеркапролом[10][11]. Це лікування стало одним з перших доступних і успішних в історії неврологічної науки, оскільки неврологи були здатні розпізнати й діагностувати захворювання, проте призначення лікування у той час було проблемою[7][12]. Перший хелатор для перорального застосування, пеніциламін, був винайдений 1956 року британським неврологом Джошем Вальше[13]. 1982 року Вальше запропонував для лікування триентин[14], а також він був першим, хто запропонував використовувати тетратіомолібдат у клініці[15]. Лікування ацетатом цинку було започатковане у Нідерландах, де лікарі Шоувінк та Хуґенраад зробили це вперше у 1961 та 1970-х роках відповідно, але цю практику надалі розвинули Брювер зі співробітниками з Мічиганського університету[16].
Спадковий характер хвороби Вільсона та її зв'язок з мутаціями у ATP7B були з'ясовані у 1980-х та 1990-х роках[17][18].
Спадковість
Ген хвороби Вільсона (ATP7B) знаходиться у 13 хромосомі (ділянка 13q14.3) і його експресія в основному проявляється у печінці, нирках та плаценті. Ген кодує ATФазу P-типу (ензим, що транспортує катіони), що транспортує мідь у жовч і включає її до складу церулоплазміну[5]. Мутації цього гену можна визначити у 90 % випадках хвороби. Більшість з них (60 %) є гомозиготами за мутацією ATP7B (дві анормальні копії), і 30 % мають лише одну анормальну копію. У десяти відсотків хворих не відмічають мутацій[19].
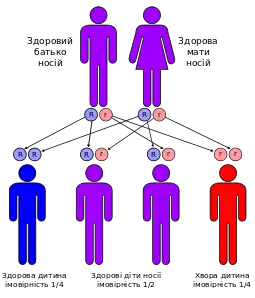
Хоча описано лише 300 випадків мутації ATP7B гену, випадки хвороби Вільсона залежать від кількості мутацій, притаманних для певної популяції. Наприклад, у західних популяціях мутація H1069Q (заміщення гістидину глутаміном у білковій позиції 1069) присутня у 37—63 % випадків, проте у Китаї ця мутація є дуже рідкісною, а мутація R778L (аргінін на лейцин у 778 позиції) трапляється набагато частіше. Достатньо мало відомо про вплив різних мутацій, хоча, згідно з деякими дослідженнями, мутації у H1069Q зазвичай відтерміновують початок симптомів[5][20].
Нормальні варіації у гені PRNP можуть модифікувати перебіг хвороби шляхом відтермінування віку початку симптомів та їх тип. Цей ген продукує PRNP (PRioN Protein), який проявляє активність у мозку та та інших органах, а також бере участь у транспортуванні міді[21]. До розвитку захворювання також може бути причетний ген ApoE, але дослідження ще не підтвердили цю теорію[20].
Захворювання успадковують за аутосомно-рецесивним типом. Для того щоб успадкувати його, обоє батьків мають бути носіями пошкодженого гену. Більшість хворих не вказують на родинну історію цією хвороби[20]. Людей з лише однією анормальною копією гену називають носіями (гетерозиготи), і у них спостерігають незначні з медичної точки зору порушення метаболізму міді[22].
Хвороба Вільсона є найпоширенішою з групи спадкових захворювань, які спричиняють накопичення міді у печінці. Усі ці захворювання можуть спричинити цироз ще у ранньому віці. До цієї групи хвороб також відносять індійський дитячий цироз (ІДЦ), тирольський ендемічний інфантильний цироз та ідіопатичний мідний токсикоз. Ці хвороби не пов'язані з мутаціями у ATP7B, наприклад, ІДЦ пов'язують із мутаціями у генах KRT8 та KRT18[20].
Патогенез
Мідь виконує багато функцій в організмі. В основному вона виступає як кофактор для деяких ферментів, таких як церулоплазмін, цитохром C-оксидаза, дофамін-β-гідроксилаза, супероксиддисмутаза і тирозиназа.
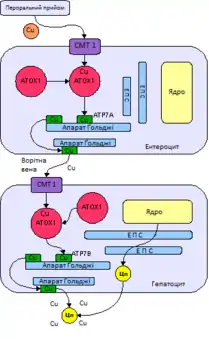
Мідь всмоктується з шлунково-кишкового тракту. Транспортний білок на клітинах тонкої кишки CMT1 (англ. Copper Membrane Transporter 1) переміщує мідь усередину клітин. Частина міді зв'язується з металотіонеїном, а інша — переміщується до комплексу Ґольджі за допомогою транспортного білка ATOX1. В апараті Ґольджі у відповідь на підвищення концентрації міді фермент ATP7A (англ. Copper-transporting ATPase 1) вивільняє цей елемент через ворітну вену у печінку. У гепатоцитах білок ATP7B зв'язує мідь з церулоплазміном і вивільняє його у кров, а також видаляє надлишок міді з жовчю. Обидві функції ATP7B порушуються при хворобі Вільсона. Мідь накопичується у тканині печінки, церулоплазмін продовжує виділятись, але з нестачею міді (апоцерулоплазмін) швидко руйнується у кров'яному руслі[20].
Коли міді у печінці стає більше, ніж білків, що її зв'язують, відбувається їх окиснювальне пошкодження за рахунок реакції Фентона. Це призводить до запалення печінки, її фіброзу, а відтак до цирозу. Також з печінки у кров'яне русло виділяється мідь, незв'язана з церулоплазміном. Ця вільна мідь осідає по всьому організму, особливо в нирках, очах і головному мозку.
Основну роль у патогенезі грає порушення обміну міді, її накопичення у нервовій (особливо уражаються базальні ядра), нирковій, печінковій тканинах і рогівці ока, а також токсичне пошкодження міддю даних органів. Порушення метаболізму міді виражається у порушенні синтезу і зниженні у крові концентрації церулоплазміну. Церулоплазмін бере участь у процесі виведення міді з організму. У печінці формується крупновузловий або змішаний цироз. У нирках у першу чергу страждають проксимальні канальці. У головному мозку уражаються більшою мірою базальні ядра, зубчасте ядро мозочка та чорна речовина. Відкладення міді в десцеметовій мембрані ока призводить до формування кілець Кейзера — Флайшера.
Гепалентикулярна дегенерація починається у дитячому або молодому віці і характеризується хронічним прогресивним перебігом. У багатьох випадках появі симптомів ураження нервової системи передують вісцеральні розлади у вигляді порушення роботи печінки і шлунково-кишкових розладів (жовтяниця, біль у правому підребер'ї, диспептичні явища). Іноді розвивається виражений гепато-спленальний синдром.
З боку нервової системи на перший план виступають екстрапірамідальні симптоми у вигляді м'язової ригідності, гіперкінезів і розладів психіки. Пірамідні симптоми можливі, але зазвичай відсутні. Чутливість, як правило, не порушується.
Розпізнають 5 форм хвороби Вільсона[23][24]:
Черевна форма — тяжке ураження печінки, що призводить до смерті ще до появи симптомів з боку нервової системи; хворіють діти. Її тривалість коливається від декількох місяців до 3—5 років.
Ригідно-аритмогіперкінетична, або рання форма відрізняється швидким перебігом; починається навіть у дитячому віці. У клінічній картині переважає м'язова ригідність, що призводить до контрактур, сповільнення рухів, хореоатетоїдних або торсійних силових рухів. Характерними є дизартрія, дисфагія, судомний сміх і плач, аффективні розлади і помірне пониження інтелекту. Захворювання триває 2—3 роки, закінчується летально.
Тремтливо-ригідна форма зустрічається частіше решти; починається в юнацькому віці, перебігає повільніше, іноді з ремісіями і раптовими погіршеннями, що супроводжуються субфебрильною температурою. Характеризується одночасним розвитком важкої ригідності й тремтіння; тремтіння є дуже ритмічним (2—8 тремтінь на секунду), різко посилюється при статичному напруженні м'язів, рухах чи хвилюванні, у стані спокою під час сну зникає. Іноді можна виявити атетоїдні хореоформні силові рухи; спостерігаються також дисфагія і дизартрія. Середня тривалість життя становить близько шести років.
Тремтлива форма починається у віці 20—30 років, перебігає досить повільно (10—15 років і довше); тремтіння різко переважає, ригідність появляється лише наприкінці хвороби, а інколи спостерігають гіпертонію м'язів; відмічають амімію, повільну монотонну мову, тяжкі зміни психіки, часто спостерігають афективні спалахи; можливі епілептиформні припадки.
Екстрапірамідно-кіркова форма зустрічається рідше решти форм. Типові для хвороби Вільсона порушення надалі ускладнюються пірамідними парезами, епілептиформними припадками і тяжким слабоумством (спостерігають обширні розм'якшення у корі великих півкуль). Триває 6—8 років, закінчується смертю.
Патологічна анатомія
У головному мозку при гепато-церебральній дистрофії розм'якшується сочевицеподібне ядро, особливо скорпула, з утворенням дрібних кіст. Уражені й інші утвори: хвостате ядро, глибокі шари кори, мозочок, а зокрема зубчасті ядра, підгорбкові ядра; в інших ділянках головного мозку зміни виражені менше.
Усі зміни поділяють на ангіотоксичні та цитотоксичні. Перші проявляються атонією судин, особливо дрібних, та зміною їхніх стінок. У результаті виникають стази, поширений периваскулярний набряк з аноксією нервової тканини та її загибеллю; часто спостерігають крововиливи та їхні сліди у вигляді скупчень гемосидерину.
Цитотоксичний компонент полягає у поширених дистрофічних змінах макроглії нервових клітин, що часто закінчується їх загибеллю. Характерним є поява глії Альцгеймера, що утворюється зі звичайних астроцитів. Нерідко зустрічають змінені нервові клітини, дуже схожі на альцгеймерську глію; схожі клітини виявляють також в печінці і нирках. В основі цих клітинних змін лежить фактор однотипного порушення клітинного обміну, імовірно, обміну нуклеїнових кислот.
Чим пізніше починається захворювання, тим повільніше воно перебігає, тим більш дифузними є зміни в головному мозку і тим більше цитотоксичний компонент переважає над ангіотоксичним. Як наслідок атрофічного цирозу, печінка є зменшеною і горбкуватою; ділянки нормальної тканини чергуються з некротичними, дегенеративними ділянками і з «острівцями» регенерації; швидке утворення судин призводить до появи анастомозів між гілками ворітної і нижньої порожнистої вен[23].
Ознаки та симптоми
Основними місцями накопичення міді є печінка та мозок, а тому захворювання печінки і нейропсихіатричні симптоми є основними ознаками, відповідно до яких ставлять діагноз[5]. Люди з проблемами із печінкою (зазвичай це діти та підлітки) звертаються за медичною допомогою частіше, ніж ті, у кого наявні неврологічні та психіатричні симптоми (зазвичай це люди у віці 20 років та старші). У декого ідентифікують хворобу лише тому, що їхнім родичам поставили цей діагноз. У багатьох пацієнтів діагностують захворювання, і виявляється, що у них були наявні симптоми, але правильний діагноз їм не могли поставити[19].
Ураження печінки
Ураження печінки може проявлятись у вигляді втомлюваності, схильності до кровотеч та психічних порушень (через печінкову енцефалопатію) та портальної гіпертензії. Остання — це стан, при якому тиск у ворітній вені є значно підвищеним, що призводить до варикозів стравохідних та інших вен, збільшення селезінки (спленомегалії) та накопичення рідини у порожнині живота (асциту). При огляді такого пацієнта спостерігають такі ознаки ураження печінки, як павукоподібні ангіоми (малі роздуті патологічні анастомози кровоносних судин, зазвичай у навколоключичній ділянці). Хронічний гепатит з високим ступенем прогресування спричинює цироз печінки ще до появи симптомів. Хоч більшість хворих на цироз є в групі підвищеного ризику гепатоцелюлярної карциноми (пухлини печінки), у хворих на хворобу Вільсона цей ризик є відносно низьким[5].
Близько 5 % усіх людей ставлять цей діагноз після виникнення у них гострої печінкової недостатності, часто у контексті гемолітичної анемії (анемія, яку виникає внаслідок руйнування еритроцитів). Це призводить до проблем в утворенні протеїнів (проявляється підвищеною коагуляцією) та порушень метаболізму у печінці. Пришвидшений метаболізм протеїнів призводить до накопичення продуктів обміну, таких як аміак, у кровоносному руслі. Коли це призводить до пошкодження мозку, то у людини розвивається хронічна печінкова енцефалопатія (психічні розлади, кома, судоми і життєво небезпечний набряк мозку)[5].
Нейропсихіатричні ознаки
Приблизно половина людей з хворобою Вільсона мають неврологічні та психіатричні прояви. У більшості відзначають як легкі когнітивні порушення, так і зміни в поведінці. Зазвичай з часом виявляють нові симптоми, часто у вигляді паркінсонізму (брадикінезія та порушення балансу при ходьбі є звичайними ознаками паркінсонізму[25]) та тремору рук, порушення експресії на обличчі, невиразної мови, атаксії (порушення координації) чи дистонії (скручувань та повторюваних рухів певної частини тіла). У деяких хворих спостерігають судоми та мігрень[5][26]. У багатьох людей присутній так званий «wing-beating»-тремор; у решти людей він відсутній, проте його можна спровокувати, попросивши хворого витягнути руки вперед[27].
Хвороба Вільсона також може вплинути на когнітивні функції. Когнітивні розлади можуть мати двоякий генез: розлад лобової частки (може проявлятись у вигляді імпульсивності, апатії, порушенням можливості планувати та ставити цілі) та субкортикальна деменція (може проявлятись у вигляді сповільнення мислення, втрати пам'яті, без ознак афазії, апраксії чи агнозії). Є теорія, що ці когнітивні порушення напряму пов'язані з психічними розладами, які спричинює хвороба[25].
До психіатричних проблем, які зумовлює хвороба Вільсона, можна віднести зміни у поведінці, депресію, тривогу та психоз[5]. Психіатричні зміни зазвичай проявляються разом із неврологічними симптомами і лише у рідкісних випадках без них. Часто ці симптоми важко визначити, або ж їм приписують інше походження. Саме через це рідко ставлять діагноз хвороби Вільсона за присутності лише психіатричних ознак[25].
Інші системи органів
Такі симптоми пов'язані з акумуляцією міді при хворобі Вільсона:
- Очі: кільця Кейзера – Флайшера (КФ кільця)[26], патогномонічна ознака, що проявляється у вигляді скупчень міді у формі кільця навколо рогівки у десцеметовій мембрані. Цю ознаку не виявляють в усіх хворих на хворобу Вільсона. Хвороба Вільсона також асоціюється із «соняшниковою катарактою», що проявляється у вигляді коричневої або зеленої пігментації передньою та задньої капсул кришталика[28]. Ці симптоми не призводять до серйозних проблем із зором[5]. Кільця Кейзера—Флайшера спостерігають у приблизно 66 % випадків хвороби (частіше у хворих з неврологічними симптомами)[19].
- Нирки: нирковий тубулярний ацидоз (другого типу), дефект реабсобції бікарбонатів у проксимальних ниркових канальцях, що призводить до нефрокальцинозу (накопичення Кальцію в нирках), ослаблення кісток (через втрату кальцію та фосфатів) і часом до аміноацидурії (екскреція незамінних амінокислот, потрібних для синтезу білків, з сечею)[5].
- Серце: кардіоміопатія (слабкість серцевого м'язу) є рідкісним симптомом хвороби Вільсона; вона може призвести до серцевої недостатності (порушення насосної функції) та аритмій (включень нерегулярного та/чи анормально швидкого чи повільного серцебиття)[5].
- Гормони: гіпопаратироїдизм (недостатність прищитоподібних залоз, що призводить до зниження рівня кальцію), безпліддя та викидні[5].
Діагностика
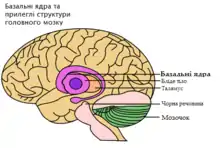
Хворобу Вільсона можна запідозрити, ґрунтуючись на будь-якому з вище перерахованих симптомів або ж у випадку, коли у близького родича діагностовано це захворювання. У більшості пацієнтів спостерігають легку форму дисфункції печінки, яку при біохімічному дослідженні підтверджує підвищений рівень білірубіну в крові, висока активність аспартатамінотрасферази та аланінамінотрансферази. При тяжких ушкодженнях печінки можна спостерігати знижений рівень альбуміну через неспроможність ушкоджених гепатоцитів синтезувати цей клас білків; також протромбіновий час (тест на зсідання крові) може бути подовженим, оскільки печінка втрачає здатність продукувати фактори зсідання крові[5]. Активність ферменту лужної фосфатази є відносно низькою у хворих із печінковою недостатністю, яку спричинює хвороба Вільсона[5]. Пацієнтам з неврологічними симптомами зазвичай проводять магнітно-резонансну томографію, що дає змогу побачити підвищену активність базальних ядер за часом спін-спінової релаксації[22]. Також МРТ може показати характерний візерунок «обличчя великої панди»[29].
Немає прямого тесту для діагностування хвороби Вільсона, проте рівень церулоплазміну та міді у крові, а також кількість міді, що виводиться з сечею за 24 години, разом допомагають скласти висновок про кількість міді в організмі. «Золотим стандартом» чи найкращим аналізом є біопсія печінки.
Церулоплазмін
Рівень церулоплазміну є анормально низьким (<0,2 г/л) у 80—95 % випадків[5]. Проте він може бути у межах норми в людей із запальними процесами в організмі, оскільки церулоплазмін є «стрес-білком». Низький рівень церулоплазміну також притаманній хворобі Менкеса та ацерулоплазмінемії, які є пов'язаними з цим явищем, проте ця ознака характерніша все-таки для хвороби Вільсона[5][22].
Поєднання неврологічних симптомів, кілець Кейзера — Флайшера та низький рівень церулоплазміну є чільними ознаками, що вказують на хворобу Вільсона. У багатьох випадках для уточнення діагнозу потрібні додаткові аналізи[22].
Мідь у крові та сечі
Рівень міді у крові є низьким, що може видатись парадоксальним, оскільки хвороба Вільсона — це захворювання надлишку міді. Однак 95 % міді у плазмі крові переносить церулоплазмін, рівень якого є звичайно низьким при хворобі Вільсона. Для діагностики також проводять забір сечі, яку зберігають у посудині, що не містить у своєму складі міді. За 24 години перевіряють рівень міді: вище 100 мкг/24год (1,6 мкмоль/24год) підтверджує діагноз хвороби, а рівень вище 40 мкг/24год (0,6 мкмоль/24год) є зоною ризику виникнення цієї хвороби[5]. Високий рівень міді в сечі не є унікальною (патогномонічною) ознакою хвороби Вільсона, натомість це явище також спостерігають при автоімунному гепатиті та холестазі (а також будь-якій іншій хворобі, що перешкоджає потраплянню жовчі у дванадцятипалу кишку)[22].
Для діагностики у дітей інколи використовують пеніциламіновий аналіз. 500-міліграмова доза пеніциламіну дитина споживає перорально, і після цього проводять забір сечі за 24 години. Рівень вище 1600 мкг (25 мкмоль) свідчить на користь хвороби Вільсона. Дорослим такий тест не проводять[22].
Біопсія печінки
Гістохімічні методи визначення рівня міді у крові чи сечі самі по собі є ненадійними і завжди потребують проведення додаткових аналізів[22]. Тож опісля підтвердження хвороби Вільсона усіма доступними тестами, вирішальним дослідженням є забір маленької частинки печінкової тканини шляхом біопсії печінки. Печінкову тканину досліджують патоморфологічно — мікроскопічним шляхом на перевірку рівня стеатозу, цирозу та кількості акумульованої міді. Рівень 250 мкг міді на грам зневодненої тканини печінки підтверджує хворобу Вільсона. Зрідка спостерігають низький рівень міді; у такому випадку сукупність даних біопсії та інших досліджень все ж дає змогу поставити діагноз гепатоцеребральної дистрофії[5].
На ранніх стадіях захворювання біопсія звичайно знаходить значний стеатоз (накопичення ліпідів у клітині), підвищений вміст глікогену в гепатоцитах та ділянки некрозу (смерті клітин). У пізніших стадіях захворювання, спостерігають зміни, схожі зі змінами при автоімунному гепатиті: інфільтрація запаленими клітинами, частковий некроз та фіброз. На останніх стадіях захворювання, як правило, біопсія виявляє цироз.
Генетичний аналіз
Аналіз мутації гену ATP7B та решти генів, пов'язаних з накопиченням міді в печінці, є варіативним при діагностиці захворювання. У випадку підтвердження мутації варто перевірити близьких родичів хворого (як мінімум покоління пробанда) на присутність прихованого захворювання[5]. Важливим фактором при аналізі геному є різниця у поширенні генів хвороби Вільсона за регіонами планети. Так, у країнах Західного світу, таких як Сполучені Штати та Велика Британія проведення генетичного аналізу може бути затрудненим, оскільки в цих країнах популяції є досить змішаними[30].
Лікування
Дієта
Загалом, рекомендують харчуватись їжею з низьким вмістом міді, уникати споживання грибів, горіхів, шоколаду, сухофруктів, печінки та молюсків[5][26].
Ліки
Ліки від хвороби Вільсона діють за двома основними механізмами: одні підвищують екскрецію міді з організму, а інші натомість зменшують всмоктування міді з продуктів харчування у кишці.
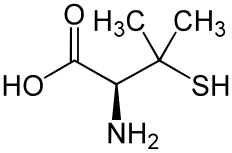
Ведучими ліками є пеніциламін[4]. Він зв'язує (хелатує) мідь, що призводить до виведення її з сечею. Таким чином, моніторинг кількості міді в сечі може бути корисним для перевірки ефективності лікувального процесу. Проте у 20 % випадків вживання пеніциламіну відзначають побічні ефекти, такі, як медикаментозний вовчак (що спричинює біль у суглобах та висип на шкірі) чи міастенію (нервовий розлад, що породжує м'язову слабкість). У хворих з неврологічною формою захворювання у половині випадків спостерігають парадоксальне погіршення симптомів. Хоча цей феномен характерний і для інших засобів лікування хвороби Вільсона, у цьому випадку зупиняють прийом пеніциламіну і призначають інші препарати[5][22]. Хворим зі стійкістю до пеніциламіну часто призначають триентину гідрохлорид, який теж має хелативні властивості. Багато лікарів рекомендують триентин як основний препарат, проте використання пеніциламіну все ж є ширшим[22]. Іншим лікрським засобом, що зв'язує мідь у кишечнику є тетраетилмолібдат, проте його використання є ще на етапі експериментального дослідження[22], але деякі дослідники вже повідомляють про його позитивний ефект[5].
bis(ethane-1%252C2-diamine)_200.svg.png.webp)
Після повернення усіх показників до норми, хворим призначають Цинк (зазвичай у вигляді препарату ацетату цинку) на заміну хелаторам для забезпечення сталого рівня міді в організмі. Цинк стимулює металотіонеїн, білок ентероцитів, що зв'язує мідь та запобігає її всмоктуванню і, відповідно, транспорту до печінки. Цинкову терапію проводять допоки не рецидивують симптоми хвороби або рівень міді в сечі не підвищиться[22].
У рідкісних випадках тяжкої неврологічної форми захворювання, коли ліки для перорального застосування не є ефективними, використовують препарат димеркаптол. Його вводять внутрішньом'язово щодекілька тижнів. Димеркаптол має низку неприємних побічних ефектів, зокрема, таких як біль у місці ін'єкції[31].
Людям з латентною формою хвороби (наприклад, тим кому діагностували хворобу через те, що їхній родич захворів) проводять загальний курс лікування, оскільки накопичення міді може принести шкоду організму у довгостроковій перспективі. Проте не є до кінця зрозумілим, якому препарату потрібно надавати перевагу в лікуванні таких хворих: пеніциламіну чи ацетату цинку[22].
Фізіотерапія
Фізіотерапія є корисною для пацієнтів з неврологічною формою захворювання. Лікування хелаторами міді може зайняти до шести місяців до початку отримання ефекту, а фізіотерапія може допомогти боротись з атаксією, дистонією та тремором, а також запобігти розвитку контрактур, що можуть перейти у дистонію[32].
Трансплантація
Трансплантація печінки є ефективним засобом для лікування хвороби Вільсона, проте використовують її лише в окремих випадках, що пов'язано з ризиком даної процедури. Як правило, її проводять людям із гострою печінковою недостатністю, що не піддається медикаментозному лікуванню або людям із розвиненою хронічною печінковою недостатністю. Трансплантацію печінки не проводять пацієнтам із тяжкими нейропсихіатричними захворюваннями, при яких ця операція не дасть вагомої користі здоров'ю[5][22].
Прогноз
За відсутності ефективного лікування хвороба Вільсона швидко прогресує і зрештою призводить до смерті. Якщо вдається вчасно діагностувати захворювання та розпочати лікування, то у більшості хворих не змінюється їх якість життя і вони мають змогу продовжувати жити повною мірою.
У тварин
Спадкові накопичення міді було описано у бедлінгтон-тер'єрів[33], у яких він впливає, як правило, тільки на печінку. Це пов'язано з мутаціями у генах COMMD1 (або MURR1)[34]. При невільсонівських захворюваннях, пов'язаних з накопиченням міді (наприклад, при дитячому індіанському цирозі), не спостерігають мутації у гені COMMD1, тож генетичний генез цих захворювань поки що неможливо визначити[35].
Див. також
Примітки
- NDF-RT
- Drug Indications Extracted from FAERS — doi:10.5281/ZENODO.1435999
- Inxight: Drugs Database
- Хвороба Вільсона - симптоми, лікування, діагностика, причини. mdovidka.com. Процитовано 14 січня 2016.
- Wilson's disease - The Lancet. www.thelancet.com. Процитовано 11 січня 2016.
- Wilson, S. a. Kinnier (1 березня 1912). Progressive Lenticular Degeneration: A Familial Nervous Disease Associated with Cirrhosis of the Liver. Brain (англ.) 34 (4). с. 295–507. ISSN 0006-8950. doi:10.1093/brain/34.4.295. Процитовано 11 січня 2016.
- WIlson's disease. Archives of Neurology 57 (2). 1 лютого 2000. с. 276–277. ISSN 0003-9942. doi:10.1001/archneur.57.2.276. Процитовано 12 січня 2016.
- Cumings, J. N. (1 грудня 1948). The Copper and Iron Content of Brain and Liver in the Normal and in Hepato-Lenticular Degeneration. Brain (англ.) 71 (4). с. 410–415. ISSN 0006-8950. doi:10.1093/brain/71.4.410. Процитовано 12 січня 2016.
- McIntyre, N.; Clink, H. M.; Levi, A. J.; Cumings, J. N.; Sherlock, Sheila (23 лютого 1967). Hemolytic Anemia in Wilson's Disease. New England Journal of Medicine 276 (8). с. 439–444. ISSN 0028-4793. PMID 6018274. doi:10.1056/NEJM196702232760804. Процитовано 12 січня 2016.
- Cumings, J. N. (1 березня 1951). The Effects of B.a.l. in Hepatolenticular Degeneration. Brain (англ.) 74 (1). с. 10–22. ISSN 0006-8950. PMID 14830662. doi:10.1093/brain/74.1.10. Процитовано 12 січня 2016.
- Denny-Brown, D.; Porter, Huntington (13 грудня 1951). The Effect of BAL (2,3-Dimercaptopropanol) on Hepatolenticular Degeneration (Wilson's Disease). New England Journal of Medicine 245 (24). с. 917–925. ISSN 0028-4793. PMID 14882450. doi:10.1056/NEJM195112132452401. Процитовано 12 січня 2016.
- Vilensky, Joel A.; Robertson, Wendy M.; Gilman, Sid (24 вересня 2002). Denny–Brown, Wilson’s disease, and BAL (British antilewisite [2,3-dimercaptopropanol]). Neurology (англ.) 59 (6). с. 914–916. ISSN 0028-3878. PMID 12297577. doi:10.1212/WNL.59.6.914. Процитовано 12 січня 2016.
- WILSON'S DISEASE - The Lancet. www.thelancet.com. Процитовано 12 січня 2016.
- TREATMENT OF WILSON'S DISEASE WITH TRIENTINE (TRIETHYLENE TETRAMINE) DIHYDROCHLORIDE - The Lancet. www.thelancet.com. Процитовано 12 січня 2016.
- Harper, P. L.; Walshe, J. M. (1 грудня 1986). Reversible Pancytopenia Secondary to Treatment with Tetrath10-Molybdate. British Journal of Haematology (англ.) 64 (4). с. 851–853. ISSN 1365-2141. doi:10.1111/j.1365-2141.1986.tb02250.x. Процитовано 12 січня 2016.
- Walshe, J. M. (1 липня 1996). Treatment of Wilson's disease: the historical background. QJM (англ.) 89 (7). с. 553–556. ISSN 1460-2725. PMID 8759497. doi:10.1093/qjmed/89.7.553. Процитовано 12 січня 2016.
- Bull, Peter C.; Thomas, Gordon R.; Rommens, Johanna M.; Forbes, John R.; Cox, Diane Wilson (1 грудня 1993). The Wilson disease gene is a putative copper transporting P–type ATPase similar to the Menkes gene. Nature Genetics (англ.) 5 (4). с. 327–337. doi:10.1038/ng1293-327. Процитовано 12 січня 2016.
- Tanzi, R. E.; Petrukhin, K.; Chernov, I.; Pellequer, J. L.; Wasco, W.; Ross, B.; Romano, D. M.; Parano, E. та ін. (1 грудня 1993). The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nature Genetics (англ.) 5 (4). с. 344–350. doi:10.1038/ng1293-344. Процитовано 12 січня 2016.
- Sign In. PMC 1856673. PMID 16709660. doi:10.1136/gut.2005.087262. Процитовано 11 січня 2016.
- Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes.
- Prion protein gene codon 129 modulates clinical course of neurological Wilson disease.
- Roberts, Eve A.; Schilsky, Michael L. (1 червня 2003). A practice guideline on Wilson disease. Hepatology (англ.) 37 (6). с. 1475–1492. ISSN 1527-3350. doi:10.1053/jhep.2003.50252. Процитовано 11 січня 2016.
- Иванова-Смоленская, И. (2006). Болезнь Вильсона-Коновалова.
- Гусев Е. И., Коновалов А. Н., Гехт А. Б. Неврология: национальное руководство. — Москва : ГЭОТАР-Медиа, 2014. — С. 553. — ISBN 978-5-9704-2890-0.
- Lorincz, Matthew T. (1 січня 2010). Neurologic Wilson's disease. Annals of the New York Academy of Sciences (англ.) 1184 (1). с. 173–187. ISSN 1749-6632. doi:10.1111/j.1749-6632.2009.05109.x. Процитовано 12 січня 2016.
- Хвороба Вільсона-Коновалова. ukrhealth.net. Процитовано 14 січня 2016.
- Pagonabarraga, J. (2012). Practical neurology. Philadelphia. с. 282. ISBN 1451142633.
- Yanoff, Myron (2008). Ophthalmology. Edinburgh. с. 411. ISBN 978-0323057516.
- Das, Shyamal K.; Ray, Kunal (1 січня 2006). Wilson's disease: an update. Nature Clinical Practice Neurology (англ.) 2 (9). с. 482–493. doi:10.1038/ncpneuro0291. Процитовано 12 січня 2016.
- Ferenci, Peter (22 червня 2006). Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing.
- Walshe, J. M. (1 липня 1996). Treatment of Wilson's disease: the historical background. QJM: An International Journal of Medicine (англ.) 89 (7). с. 553–556. ISSN 1460-2725. PMID 8759497. doi:10.1093/qjmed/89.7.553. Процитовано 14 січня 2016.
- Wilson's disease: clinical management and therapy - Journal of Hepatology. www.journal-of-hepatology.eu. Процитовано 13 січня 2016.
- Sternlieb, I; Twedt, D C; Johnson, G F; Gilbertson, S; Korotkin, E; Quintana, N; Scheinberg, I H (1 січня 1977). Inherited copper toxicity of the liver in Bedlington terriers.. Proceedings of the Royal Society of Medicine 70 (Suppl 3). с. 8–9. ISSN 0035-9157. PMC 1543595. PMID 122681. Процитовано 12 січня 2016.
- Sluis, Bart van de; Rothuizen, Jan; Pearson, Peter L.; Oost, Bernard A. van; Wijmenga, Cisca (15 січня 2002). Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Human Molecular Genetics (англ.) 11 (2). с. 165–173. ISSN 0964-6906. PMID 11809725. doi:10.1093/hmg/11.2.165. Процитовано 12 січня 2016.
- The canine copper toxicosis gene MURR1 does not cause non-Wilsonian hepatic copper toxicosis - Journal of Hepatology. www.journal-of-hepatology.eu. Процитовано 12 січня 2016.
Джерела
- Richard K Gilroy, Rahil Shah, Michael H Piper Wilson Disease. Updated: Jan 11, 2016 Medscape. Pediatrics, Neurology & Neurosurgery / Chief Editor: Julian Katz
- МОЗУ, НАКАЗ від 26 липня 2016 року N 769 «Про затвердження та впровадження медико-технологічних документів зі стандартизації медичної допомоги при хворобі Вільсона» Ел.джерело
Посилання
- Міжнародна асоціація щодо хвороби Вільсона
- Хвороба Вільсона UK
- Реєстр випадків хвороби Вільсона (Організація геному людини)
- Хвороба Вільсона на сайті mdovidka
- Хвороба Вільсона на сайті ukrhealth.net
|
|
Ця стаття належить до добрих статей української Вікіпедії. |
