Флуоресценція в біологічних дослідженнях
Флуоресценція знайшла широке застосування у різноманітних прикладних біологічних та біомедичних дослідженнях.[1] Це фізичне явище, суть якого полягає в короткочасному поглинанні кванта світла флуорофором (речовиною, що здатна флуоресціювати) із наступною швидкою емісією іншого кванту, що має властивості, відмінні від вихідного.[2] Багато напрямків у біофізиці, молекулярній та клітинній біології виникли та розвиваються саме завдяки впровадженню нових методів, що базуються на флуоресценції. Варто навести декілька прикладів.
Для біофізиків флуоресценція стала швидким та чутливим методом дослідження структури, динаміки та функцій біологічних макромолекул — нуклеїнових кислот[3] та білків.[4]
Метод секвенування ДНК за Сангером був значно вдосконалений у другій половині 1980-х років саме завдяки впровадженню флуоресцентної детекції. Важливим наслідком цього стала вища швидкість та надійність секвенування. Окрім цього метод було автоматизовано.[5][6] Це відкрило технічну можливість проведення широкомасштабного (за масштабами того часу) секвенування та дозволило розпочати проект «Геном людини» на початку 1990-х років. Крім прямого секвенсування (методом Сангера), флуоресценція продовжує використовуватись у методах секвенування ДНК наступних поколінь (англ. Next generation sequencing).[7][8]
Флуоресценція дала новий поштовх для розвитку клітинної біології. Завдяки конфокальній флуоресцентній мікроскопії та розробці нових флуоресцентних міток на базі зеленого флуоресцентного білка (GFP) та його аналогів з'явилась можливість отримувати специфічні контрастні забарвлення та робити фотознімки з високим розділенням багатьох внутрішньоклітинних білкових структур. Розробка нових флуоресцентних зондів — речовин що змінюють флуоресценцію коли до них приєднується певна молекула — дала можливість детально досліджувати хімічних склад живих клітин та навіть організмів, а також його зміни у часі і просторі, що поклало початок флуоресцентній молекулярній візуалізації (англ. molecular imaging).[9][10]
Починаючи із середини XX-го століття, аналітичні методи, що базуються на використання явища флуоресценції, широко застосовуються у клінічній хімії та молекулярній діагностиці. Зокрема були розроблені та впроваджені чутливі методи для швидкого аналізу стероїдних гормонів, порфіринів, катехоламінів, метаболітів медичних препаратів та інших діагностично важливих хімічних речовин у сечі та плазмі крові.[11] За допомогою імуноферментного аналізу (ELISA) із використанням флуорогенних субстратів проводять детекцію біомаркерів різних захворювань.
Активно розроблюються методи флуоресцентної діагностики in vivo.[12][13] Зокрема створені флуоресцентні зонди, що селективно забарвлюють злоякісні утворення та допомагають виявляти їх під час ендоскопічного обстеження або томографії[14]. Також на основі флуоресцентного забарвлення тканин були розроблені новітні методики проведення хірургічних операцій для видалення злоякісних пухлин (англ.: image-guided surgery). Перед операцією ракова пухлина селективно забарвлюється флуоресцентним барвником. Під час самої операції спеціальне обладнання реєструє флуоресцентний сигнал, дозволяючи хірургу більш точно розрізняти злоякісну та здорову тканину.[15][16]
Фізичні основи флуоресценції
Флуоресценція — це одне з явищ, які можуть відбуватись під час взаємодії електромагнітного випромінювання з речовиною.
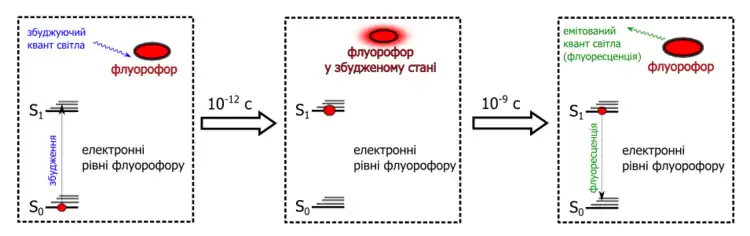
За нормальних умов переважна більшість молекул перебуває в основному електронному стані. Молекули можуть поглинати кванти електромагнітного випромінювання певної енергії та переходити у збуджений стан. Це відповідає переходу одного електрону з найвищої зайнятої на найнижчу вільну молекулярну орбіталь (ВЗМО → НВМО). Відповідно до їх мультиплетності, основний та збуджений електронні стани позначають та . Збудження більшості флуорофорів () відбувається під дією короткохвильового ультрафіолетового (довжина хвилі 300—400 нм) або видимого (довжина хвилі 400—800 нм) світла. Після переходу флуорофору у збуджений стан відбувається релаксація — процес при якому молекула втрачає частину енергії; при цьому вона опускається до найнижчого коливального підрівню електронного рівню . У рідкому середовищі за нормальних умов цей процес відбувається за час порядку декількох пікосекунд (10−12 с). Згідно з правилом Каші, саме з найнижчого коливального підрівню електронного рівню відбувається перехід в основний електронний стан, що супроводжується флуоресценцією. Через втрати енергії під час релаксації та з деяких інших причин флуоресцентне випромінення має меншу енергію (та відповідно більшу довжину хвилі) порівняно зі світлом, що поглинається під час збудження.[2]




На малюнку, що наведений вище, не показані інші процеси, які конкурують із флуоресценцією. Зокрема, за час існування молекули у збудженому стані може відбутись внутрішня конверсія, тобто безвипромінювальний перехід . Існує також можливість переходу молекули у триплетний стан за рахунок інтеркомбінаційної конверсії. Повну картину можливих переходів можна побачити на діаграмі Яблонського. Флуоресценцію не слід плутати з іншими типами люмінесценції, такими як фосфоресценція, Хемолюмінесценція, біолюмінесценція тощо.[17]


Детекція флуоресценції
У найпростішому випадку для спостереження флуоресценції потрібні лише розчин флуоресцентної сполуки та відповідне джерело світла для збудження. Зручним джерелом збудження є ручні ультрафіолетові лампи.
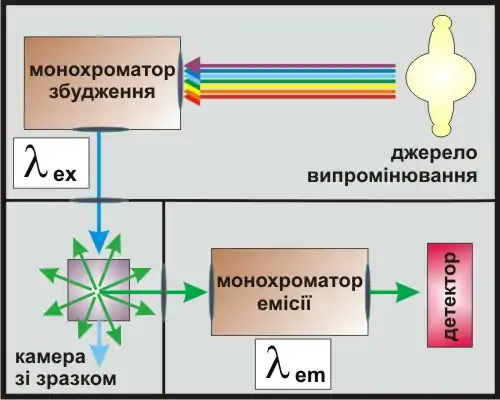
Основними приладами для вивчення флуоресценції в лабораторних умовах є спектрофлуориметри.[18] Сильно спрощена схема такого приладу показана на малюнку.
У спектрофлуориметрах використовують різні джерела збудного світла, найчастіше ксенонові лампи високого тиску. Вони дають широкий спектр емісії (від ультрафіолетового до інфрачервоного світла). Випромінювання від лампи надходить до монохроматору збудження, який дає на виході світло з певною потрібною довжиною хвилі . Після цього монохроматичний промінь направляється на зразок, спричиняючи його флуоресценцію. Важливим є те, що флуоресцентна емісія ізотропна, тобто її інтенсивність однакова по всіх напрямках (не залежить від кута під яким вона спостерігається). Це дає можливість легко відділити її від збудного світла. Розташування детектора під кутом 90° до напрямку збудження дає можливість вловлювати виключно ті фотони, які є результатом флуоресцентної емісії. Флуоресцентне світло пропускається крізь ще один монохроматор (монохроматор емісії, ). Після цього інтенсивність світлового потоку вимірюється за допомогою детектору.


Перераховані тут компоненти (джерело світла, монохроматори, детектор) у різних варіантах і комбінаціях присутні у всіх приладах, що вимірюють флуоресценцію. Залежно від призначення приладу, конфігурація та характеристики кожного з елементів системи можуть змінюватись. Наприклад, замість монохроматорів можуть використовуватись набори світлофільтрів; як джерела монохроматичного світла можуть використовуватись лазери[1].
Характеристики флуоресцентної емісії
Існує ряд кількісних параметрів, що описують флуоресцентне випромінення.
| Параметр | Позначення | Опис |
|---|---|---|
| Інтенсивність флуоресценції | , або | Цей параметр пропорційний кількості фотонів, які досягають детектора протягом одиниці часу. Вона вимірюється в кількості фотонів за секунду (cps, counts per second), або у відносних одиницях (A.U., arbitrary units). Цей параметр залежить як від досліджуваного зразку, так і від приладу, на якому здійснюють виміри. |
| Спектр флуоресценції | Спектр флуоресценції — це залежність інтенсивності флуоресценції від довжини хвилі детекції (λem), записана за сталого значення довжини хвилі збудження (λex). У найпростішому випадку залежність має вигляд асиметричної кривої з одним максимумом. Позиція максимуму емісії показує, яким кольором флуоресціює сполука. Так, максимум флуоресценції при 450 нм приблизно відповідає синьому світлу, максимум при 650 нм означає червону флуоресценцію, тощо. Ширина спектру флуоресценції органічних барвників складає від кількох десятків до кількох сотень нанометрів. Ширина спектру флуоресценції квантових точок є меншою і складає кілька десятків нанометрів. | |
| Час життя флуоресценції | Для кожної флуоресцентної сполуки можна виміряти величину, що має розмірність часу та має назву «час життя флуоресценції». Це величина порядку наносекунд для більшості флуорофорів. Вона показує усереднений час існування молекули у збудженому стані. Якщо побудувати графік затухання флуоресценції у часі, він буде мати вигляд експоненційної кривої. У найпростішому випадку спадання буде моноекспоненційним, тобто буде мати вигляд прямої у логарифмічних координатах.
 Час життя в 1 нс не означає, що флуоресценція зразку повністю зникне за одну наносекунду. Ця величина означає середній час існування збудженого флуорофору у великій популяції. Флуоресценція, подібно до радіоактивного розпаду, є стохастичним процесом. Тобто, кожна окремо взята молекула може випромінити фотон за час як коротший, так і довший [1]. | |
| Квантовий вихід флуоресценції | Показує, яка частка поглинутих фотонів випромінюється у вигляді флуоресценції. Як зазначалось вище, флуоресценція конкурує з рядом інших процесів, такими як внутрішня та інтеркомбінаційна конверсія. Окрім них існує також можливість руйнування флуорофору шляхом хімічного перетворення (фотознебарвлення). Внаслідок цього кількість фотонів, що випромінюються внаслідок флуоресценції, завжди нижча ніж кількість поглинутих фотонів (Ф < 1). |





Суміжні явища, важливі для біологічних застосувань
У дослідженнях біологічних систем корисними є деякі явища, пов'язані з флуоресценцією.
| Явище | Опис та основні характеристики |
|---|---|
| Анізотропія флуоресценції | Показує, наскільки змінюється орієнтація збуджених молекул за час існування збудженого стану.[1][17] Для вимірювання анізотропії флуоресценції необхідні спектрофлуориметри, що обладнані поляризаторами збудного та флуоресцентного світла. Знаючи інтенсивність флуоресцентного світла, поляризованого у паралельній та перпендикулярній площинах, анізотропію можна розрахувати за формулою:
Анізотропія флуоресценції показує наскільки вільно обертається молекула за час існування збудженого стану. Вільні флуорофори у рідких розчинниках за нормальних умов обертаються швидко, що викликає повну деполяризацію (r = 0). Якщо флуорофор зв'язаний із великою біомолекулою, наприклад із білковою глобулою, такий комплекс обертається в просторі повільніше, що призводить до ненульових значень анізотропії. Теоретично можливий максимум дорівнює 0.4[17]. Анізотропія флуоресценції широко використовується для вивчення білків та взаємодій між ними[19]. |
| Гасіння флуоресценції | Інколи інтенсивність флуоресценції суттєво зменшується за наявності певних сполук у розчині. Таке явище називається гасінням флуоресценції, а сполуки що його викликають — гасниками (англ. quencher) 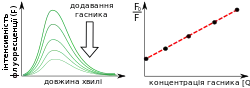 Класичним прикладом є кисень, який є гасником для великої кількості органічних флуорофорів[1]. Математично гасіння флуоресценції описується рівнянням Штерна-Вольмера. Тут — відношення початкової флуоресценції до флуоресценції в присутності гасника; — константа Штерна-Вольмера; — концентрація гасника. Графік у координатах — називається графіком Штерна-Вольмера. Гасіння буває статичним і динамічним. При статичному гасінні флуорофор у основному електронному стані утворює нефлуоресцентний комплекс із гасником. При динамічному гасінні утворюється звичайний збуджений стан, який руйнується гасником ще до того, як встигає відбутись флуоресцентна емісія[1]. Експерименти з гасінням флуоресценції триптофану акриламідом часто використовуються для аналізу конформації білкових молекул[1]. Окрім того гасіння органічних флуорофорів використовується при розробці флуоресцентних зондів.[17] |
| FRET — Ферстерівський резонансний перенос енергії | Ферстерівський резонансний перенос енергії (англ.: Förster resonance energy transfer, FRET) — процес у якому бере участь два флуорофори, донор (D) і акцептор (A) переносу. Під час FRET відбувається перенесення енергії від одного флуорофору до іншого. Тобто збуджуючи одну молекулу (донор), можна спостерігати флуоресценцію він іншої (акцептору).
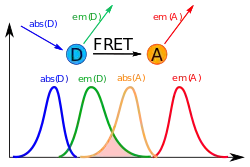 Fret — emission spectra scheme.svg Для FRET необхідно дотримання декількох умов[1], а саме:
Важливим є те, що FRET відбувається на відстанях, сумірних із розмірами біологічних об'єктів, таких як білкові глобули або мембрани клітин. При цьому відносна ефективність переносу енергії обернено залежить від відстані між FRET-партнерами. Ефективність FRET розраховується за формулою.
тут є Ферстерівським радіусом: такою відстанню між донором та акцептором, при якій ефективність переносу дорівнює ½. Якщо дві біомолекули, мічені FRET-парою знаходяться на великій відстані, при збудженні донора буде спостерігатись тільки його власна флуоресценція. У випадку коли молекули зближені у просторі, при збудженні донора буде спостерігатись емісія акцептора.  Через свою залежність від відстані, FRET став своєрідною «молекулярною лінійкою», яка дозволяє вимірювати відстані між молекулами, кожна з яких мічена одним із партнерів переносу. FRET може спостерігатись між різними за своєю хімічною природою флуорофорами; можливе також використання цього явища на рівні окремих молекул[20] або у час-розділеному форматі (англ. time-resolved FRET), що дає додаткову інформацію про динаміку та гетерогенність складних молекулярних системи[21][22]. |







Переваги флуоресцентних методів дослідження

Надвисока чутливість
Важливим показником методу детекції, що базується на певному явищі, є його чутливість — те, яка кількість вихідного матеріалу достатня для його однозначної ідентифікації. За своєю чутливістю флуоресценція є абсолютним рекордсменом, перевершуючи методи детекції, що базуються на поглинанні світла або використанні радіоактивного розпаду.[1] Сучасні інструменти можуть ідентифікувати окремі флуоресцентні молекули. Це сприяло розвитку окремого напрямку — одномолекулярної флуоресцентної спектроскопії (ОФС, англ.: Single Molecule Fluorescence Spectroscopy).[23][24] Одномолекулярна флуоресцентна спектроскопія відкрила нові можливості для вивчення біологічних систем на молекулярному рівні. Наприклад, класичні методи дослідження біомолекул, такі як спектроскопія ядерного магнітного резонансу, мають справу з великими зразками, що містять велику кількість молекул, тому весь час доводиться мати справу з усередненим сигналом. Інші методи, такі як електронна мікроскопія, дозволяють фізично спостерігати за окремими молекулами; проте такі методи не дають можливість вивчати їх у біологічно-релевантних умовах. Одномолекулярна флуоресцентна спектроскопія стала важливим методом дослідження, що поєднує можливість спостерігати за окремими молекулами з можливістю досліджувати їх у динаміці та за біологічно-релевантних умов. Наприклад, саме завдяки ОФС стало можливим вивчати згортання та динаміку білків та ДНК на рівні окремих молекул.[25][26][27][20] Також на основі ОФС були створені методи для секвенування окремих молекул ДНК та для спостереження за окремими флуоресцентними молекулами у клітині з використанням флуоресцентної мікроскопії надвисокої роздільної здатності.[28][29] За допомогою сучасних методів одномолекулярної мікроскопії вдається не тільки визначити положення окремих молекул у клітині з точністю до декількох десятків нанометрів (що значно перевершує можливості традиційної світлової мікроскопії), але і слідкувати за їх перетвореннями у реальному часі.[30]
Мультиплексність детекції
Існує велика кількість флуорофорів, кожен з яких характеризується певним максимумом емісії (кольором флуоресценції). Це відкриває можливість для мультиплексної детекції, тобто для спостереження за декількома об'єктами одночасно, якщо вони закодовані флуорофорами з різними кольорами емісії. Спектри емісії флуорофорів повинні при цьому не перекриватись. Якщо використовувати флуорофори з вузькими спектрами, такі як квантові точки, можливо спостерігати навіть за п'ятьма внутрішньоклітинними цілями одночасно.[31]

Сумісність із живими організмами
Існує можливість проводити дослідження із використанням флуоресценції на живих клітинах і навіть цілих організмах.[10] Видиме флуоресцентне світло не поглинається біологічними макромолекулами, водою та іншими компонентами живих клітин та не впливає на процеси, що відбуваються в клітині.
За останні роки були розроблені численні біосумісні флуорофори та флуоресцентні зонди. Серед них особливо виділяються флуоресцентні білки. Завдяки генній інженерії флуоресцентні білкові маркери різних кольорів можуть бути приєднані до протеїнів у різних лабораторних організмах (fusion proteins). На фотографії праворуч зображені миші, у геном яких був вбудований ген eGFP — підсиленого зеленого флуоресцентного білку.
При візуалізації флуоресценції в живих тканинах певну проблему складає поглинання світла з короткими довжинами хвиль. У зв'язку з цим широку популярність як лабораторний організм здобула Danio rerio — маленька акваріумна рибка, яка є повністю прозорою для видимого світла на перших етапах розвитку. Це робить її зручним модельним організмом для досліджень із використанням флуоресцентних міток та зондів[32].

Висока швидкість відповіді
Флуоресценція є дуже швидким процесом, що відбувається в наносекундній шкалі часу (у випадку окремих комплексів металів — у мікросекундній). За секунду одна молекула флуорофору може випроменити мільйони фотонів, кожен з яких містить інформацію про оточення, в якому перебувала молекула безпосередньо перед емісією.[17] Завдяки цьому флуоресценцію зручно використовувати для дослідження швидких процесів, таких як згортання та динаміка окремих білкових молекул.[26]
Високе просторове розділення
Просторове розділення методу вказує, на якій мінімальній відстані повинні знаходитись об'єкти для того, щоб їх можна було однозначно розрізнити. Просторове розділення дуже важливе у дослідженнях живих систем на мікроскопічному рівні. Лінійний розмір окремих клітинних структур, таких як, наприклад, ядерні пори, може складати десятки нанометрів, що робить їх недосяжними для класичної оптичної мікроскопії.
Завдяки деяким особливостям процесу флуоресценції, таким як, наприклад, можливості керовано позбавлятися небажаної емісії на певних ділянках зразку за допомогою додаткового опромінення (stimulated emission depletion) наприкінці XX століття були розроблені методи оптичної мікроскопії надвисокої роздільної здатності (super resolution microscopy).
Якщо для конфокальної флуоресцентної мікроскопії максимально досяжне просторове розділення становить близько 200 нм, для методів мікроскопії надвисокої роздільної здатності (STED, PALM тощо) роздільна здатність досягає кількох десятків нанометрів.[33][34]
Флуорофори
Здатність флуоресціювати притаманна далеко не всім хімічним сполукам. Наприклад, з-поміж двадцяти протеїногенних амінокислот флуоресцентні властивості мають лише три: фенілаланін, тирозин, та триптофан.[35] Флуоресценція останнього широко використовується для вивчення конформацій, динаміки та взаємодій триптофанвмісних білків.[35][1] Пуринові та піримідинові азотисті основи, що входять до складу ДНК та РНК майже не флуоресціюють за нормальних умов.[36]
Існує велике розмаїття штучних флуоресцентних сполук із різними фотофізичними властивостями.[37][38][39][40] Основними класами є малі органічні барвники, координаційні сполуки лантаноїдів, флуоресцентні білки та напівпровідникові нанокристали. Кожен клас має свої специфічні особливості, переваги та недоліки.
Малі органічні флуорофори
Малі органічні флуорфори є найбільшим класом флуоресцентних сполук. У більшості випадків це відносно невеликі органічні речовини, що містять декілька ароматичних фрагментів. Молекулярна маса більшості органічних флуорофорів є меншою одного кілодальтона.[17]
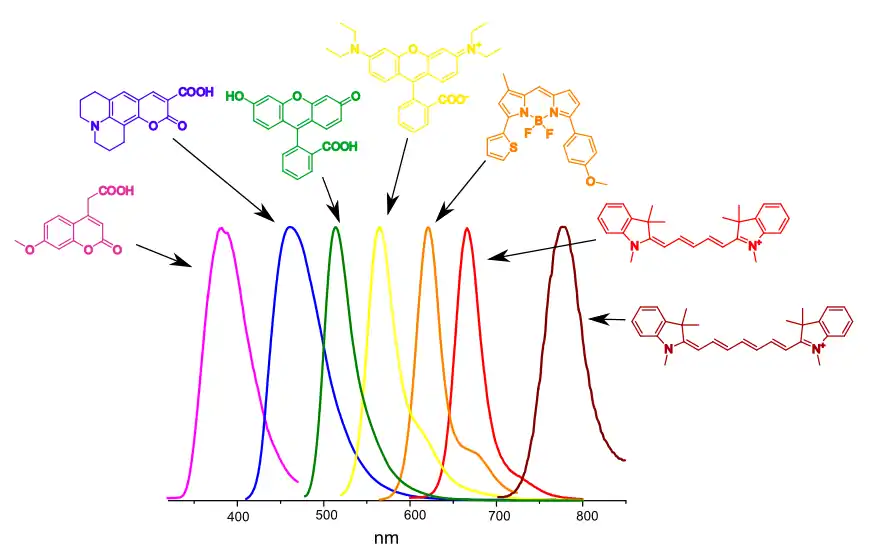
Флуоресцентними властивостями володіє надзвичайно велика кількість органічних сполук. Практичне значення має лише їх обмежена кількість, — похідні декількох базових структур.[41] Це похідні кумарину, флуоресцеїну,[42] родаміну,[42][43] бор-дипірометену BODIPY,[44] ціанінові[45] та скваринові[46] барвники.
Колір флуоресценції малих органічних барвників може змінюватись у дуже широких межах. Так, наприклад, похідні кумарину та флуоресцеїну мають синю та зелену флуоресценцію, відповідно. Похідні родаміну та BODIPY можуть мати жовто-червону флуоресценцію, тоді як на базі ціанінів та скваринів створені барвники що флуоресціюють у червоному та ближньому інфрачервоному кольорі. Флуоресценцію барвника можна керувати, змінюючи хімічну природу функціональних груп, що приєднані до флуорофору[41].
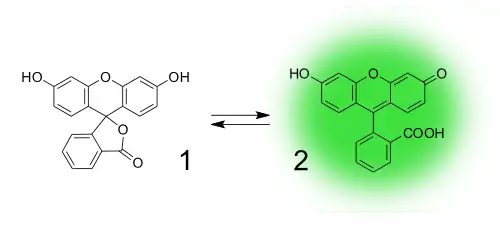
Іншою особливістю малих органічних флуорофорів є те, що їх флуоресценцію можна «вмикати» за допомогою мінімальних змін у хімічній структурі. Це широко використовуються в створенні флуоресцентних зондів на основі таких молекул. Прикладом є флуоресцеїн, який може існувати у формі двох таутомерів: нефлуоресцентній лактонній формі та флуоресцентній відкритій формі.
Нефлуоресцентні сполуки на основі «закритої» форми здатні перетворюватись на флуоресцентні під дією певних хімічних речовин.[47]
Існують хімічні методи для вимкнення та увімкнення флуоресценції інших флуорофорів[48]. Подібні реагенти, що збільшують флуоресценцію в певних умовах, називають «флуорогенними зондами».[49]
Значною перевагою органічних флуорофорів є можливість змінювати їх фотофізичні властивості за допомогою варіювання функціональних груп. Іншими перевагами є малий розмір та можливість селективного ковалентного мічення біомолекул. Недоліками є помірні квантові виходи флуоресценції, низька яскравість, низька хімічна стабільність та швидке фотознебарвлення під дією лазерного випромінення[17].
Нещодавно розроблені нові покоління флуоресцентних барвників із покращеними властивостями.[50][51] В дослідженнях нових флуорофорів використовуються сучасні методи органічної хімії, такі як комбінаторний органічний синтез.[52]
Координаційні сполуки
Флуоресцентні властивості притаманні деяким катіонам металів із групи лантаноїдів (Ln3+). За поглинання і випромінювання світла цими атомами відповідають переходи електронів f-підрівня, які в більшості випадків є квантовомеханічно забороненими.[17] Через це флуоресценція лантаноїдів має певні особливості, зокрема дуже довгі часи життя збудженого стану, які що на 3-4 порядки вище ніж часи життя органічних флуорофорів. Через наявність декількох можливих електронних переходів із різними енергіями, у спектрах флуоресценції лантаноїдів спостерігається набір окремих смуг, характерних для кожного елементу. Колір емісії може змінюватись від блакитного (Tm) до інфрачервоного (Er).[53] Зазвичай лантаноїди використовують у формі комплексів з органічними лігандами, які підвищують ефективність збудження атомів металу (сенсибілізація).

Флуоресцентні білки
Важливою групою флуорофорів є флуоресцентні білки (англ.: fluorescent proteins). Перший представник цього класу — зелений флюоресцентний білок (GFP) — був виділений із медузи Aequorea victoria в 1962 році.[54] Це відносно невеликий білок із молекулярною масою 27 кДа, що поглинає синє світло та флуоресціює зеленим.
У 1996-му році тривимірна будова дикого GFP та його мутантів була досліджена методом дифракції рентгенівських променів.[55][56] Було з'ясовано, що білок має структуру подібну до циліндру, який утвореного декількома бета-листами. У центрі циліндру розташований флуорофор, який утворюється завдяки хімічній реакції між амінокислотними залишками серину, тирозину та гліцину (амінокислоти 65-67).
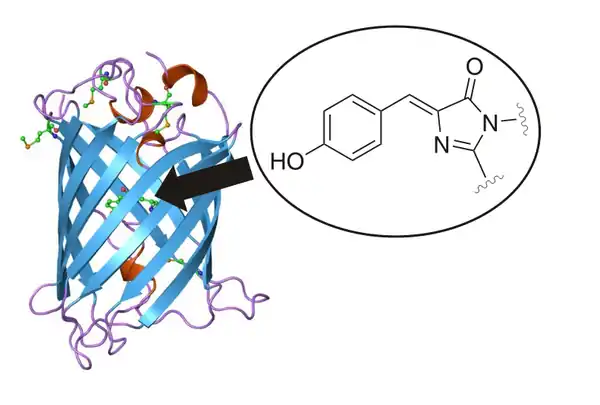
Дослідження показали, що згортання поліпептидного ланцюгу GFP у циліндричну структуру та утворення функціонального флуорофору є спонтанним процесом та не вимагає ніяких посттрансляційних модифікацій або кофакторів окрім молекулярного кисню.[57][58] Як наслідок, завдяки методами генної інженерії GFP можна успішно експресувати в багатьох організмах, які природно не мають флуоресцентних білків.

Також була розроблена технологія використання GFP як маркерного протеїну, який можна приєднати до іншого внутрішньоклітинного білку. Якщо поєднати ген GFP із геном, що кодує певний білок, після транскрипції та трансляції утвориться новий гібридний протеїн, що складається з двох частин. При цьому GFP-частина самостійно перетвориться на компактну циліндричну структуру із флуорофором всередині. Оскільки GFP є відносно невеликим біохімічно інертним білком, він із високою ймовірністю не буде заважати своєму «партнеру» виконувати свої функції в клітині. Але при цьому вся гібридна конструкція буде яскраво флуоресціювати, що дасть можливість спостерігати за її виникненням та переміщеннями.[57] Наприклад, якщо ввести ген GFP у гени, що кодують білки цитоскелету, останній стане яскраво флуоресціювати.[59]
Заміною окремих амінокислот у дикому GFP методом точкового мутагенезу були отримані флуоресцентні білки з покращеними властивостями. Наприклад, зміна окремих амінокислот з оточення флуорофору дала мутанти з іншими кольорами флуоресценції (синім, жовтим, червоним та інфрачервоним).[60][61] Також вдалося отримати варіанти GFP із меншим часом утворення флуоресцентної форми (дозріванням), з вищою фотостійкістю та вищими квантовими виходами флуоресценції.
Розроблені фотоактиваційні флуоресцентні білки (англ. photoswitchable fluorescent proteins), які можна «вмикати» та «вимикати» опроміненням світлом певного кольору.[62][63]
На базі флуоресцентних білків були розроблені генетично програмовані флуоресцентні сенсори.[64][65] Окрім цього флуоресцентні білки знайшли широке застосування у флуоресцентній спектроскопії надвисокої роздільної здатності.[66]
Революційний вплив флуоресцентних білків на сучасну біологію та біотехнологію було відзначено Нобелівською премією з хімії 2008 року, яка була вручена Осаму Шимомурі, Мартіну Шалфі та Роджеру Тсьєну.[67]
Флуоресцентні наночастинки та нанокластери
Іншою групою флуоресцентних сполук є напівпровідникові нанокристали, або квантові точки (англ. Quantum dots). При зменшенні фізичних розмірів частинки напівпровідника до нанометрових розмірів вони починають проявляти властивості, відмінні від об'ємних напівпровідників. Зокрема мова іде про квантові ефекти. При взаємодії квантової точки з електромагнітним випромінюванням утворюється екситон, що замкнений у потенційній ямі. Рекомбінація екситону призводить до вивільнення енергії. Завдяки цьому частинки нанометрових розмірів утворені з таких напівпровідникових речовин як селенід кадмію, здатні поглинати світло та флуоресціювати.[17]

Через відмінність у хімічній будові та природі основного та збудженого електронного стану, фотофізичні властивості квантових точок відрізняються від властивостей органічних флуорофорів та флуоресцентних білків. По-перше, квантові точки дають вузький та симетричний спектр емісії, положення максимуму якого залежить від діаметра квантової точки та матеріалу, з якого вона утворена. Якщо варіювати концентрацію реагентів при синтезі, можна досягти формування квантових точок переважно одного діаметра, які будуть мати свій специфічний колір флуоресценції. Так, наприклад, для CdSe зміна розміру ядра від 13 до 24 нанометрів призводить до зміни флуоресценції від блакитної (λem = 500 нм) до червоної (λem = 610 нм).[17] Важливим є те, що вигляд спектру збудження флуоресценції не залежить від діаметра; це означає що можна досягти одночасного збудження багатьох різних типів квантових точок використовуючи лише одну хвилю збудження, що дуже зручно для використання у флуоресцентній мікроскопії.
Іншими перевагами квантових точок над органічними флуоресцентними барвниками є високі квантові виходи флуоресценції та висока стійкість до фотознебарвлення.[17]
У той же час квантові точки мають і ряд недоліків. По-перше, це великі фізичні розміри, що перевищують величину більшості біологічних молекул. По-друге, матеріали з яких виготовляються квантові точки (Cd, Pb, Se, Hg), є дуже токсичними для живих клітин і організмів.[68] Для зменшення токсичності застосовується багатоступеневий дизайн квантових точок. Напівпровідникове ядро (core) покривається подвійною захисною оболонкою зі спорідненого матеріалу (для селеніду кадмію таким матеріалом є сульфід цинку) та гідрофільною полімерною оболонкою, яка збільшує розчинність квантової точки у водному середовищі та дає можливість хімічно прив'язувати до поверхні інші молекули.[69]
Квантові точки широко використовуються у флуоресцентній мікроскопії та молекулярній діагностиці in vitro[70]; також розробляються методи для використання їх у молекулярному іміджингу та діагностиці in vivo.
Окрім квантових точок існують інші флуоресцентні частинки нанометрових розмірів. Прикладом є кремнієві наночастинки з ковалентно прив'язаними до поверхні органічними барвниками[71] які мають суттєво нижчу токсичність порівняно із квантовими точками. Відомі також наночастинки, утворені з полімерних органічних сполук.[72] Іншим прикладом є золоті та срібні нанокластери синтезовані на матриці з ДНК, які демонструють флуоресцентні властивості, складаючись при цьому всього з кількох атомів металу.[73][74]
Флуоресцентні зонди та мітки
Флуоресцентні речовини, що застосовуються в біології, можна умовно поділити на дві великі групи: флуоресцентні зонди (англ. fluorescent probes) та флуоресцентні мітки (англ. fluorescent tags, fluorescent tracers).[17][75]
- Флуоресцентні мітки слугують для того, щоб ідентифікувати наявність або просторове положення молекули, що досліджується. Флуоресцентна мітка має бути хімічно стабільною і демонструвати стабільну флуоресценцію, яка не залежить від зовнішніх факторів і мінімально змінюється в часі. Таким чином вона діє як пасивний «маяк», який сигналізує про місце знаходження молекули, до якої він прив'язаний.
- Флуоресцентний зонд є більш складним за своїми функціями. Це молекулярна конструкція, яка може існувати у двох станах: «вимкненому» і «увімкненому». Ці стани розрізняються між собою певними параметрами флуоресцентної емісії (найчастіше квантовим виходом флуоресценції, позицією максимуму в спектрі емісії або часом життя збудженого стану). Перехід між «увімкненим» та «вимкненим» станами залежить від наявності в середовищі зонду тих молекул, які він повинен розпізнавати.
Флуоресцентні мітки
Найпоширенішими флуоресцентними мітками в клітинній та молекулярній біології є флуоресцентні білки. Мічення зеленим флуоресцентним білком (GFP-tagging) та його аналогами є рутинною процедурою, що використовуються при вивченні структури та функцій білків у різних модельних організмах. У базі даних Pubmed нараховуються десятки тисяч статей, що містять ключові слова «GFP», «GFP-tagging», тощо.
У наш час завдяки поєднанню технологій селективного GFP-мічення білків, високопродуктивної автоматичної мікроскопії та комп'ютерного аналізу зображень можливе паралельне вивчення локалізації та функцій сотень різних білків.[76]
Флуоресцентні білки неможливо прямо використовувати для ковалентного мічення нуклеїнових кислот. Для того щоб досліджувати ДНК та РНК за допомогою флуоресцентних протеїнових маркерів, використовують такий підхід. У ланцюг нуклеїнової кислоти вводять послідовність, з якою селективно зв'язується певний протеїн (репресор, фактор транскрипції, тощо). Сам протеїн мітять потрібним флуоресцентним білком. Флуоресцентно-марковані білки споріднені до ДНК та РНК зв'язуються зі своїми мішенями, показуючи їх просторову локалізацію. Практичними реалізаціями цієї стратегії є візуалізація ДНК в еукаріотичних клітинах за допомогою GFP-lac-репрессора[77], для візуалізації РНК за допомогою λN системи, тощо.[78]
Флуоресцентні зонди
Відповідно до своєї назви, флуоресцентний зонд має за мету передавати досліднику інформацію про середовище, в якому він знаходиться. Флуоресцентним зондом називається молекулярна конструкція, що змінює один із параметрів флуоресценції (інтенсивність, час життя, максимум спектру флуоресценції), коли зв'язується зі своєю мішенню. Флуоресцентні зонди є зручним інструментом для візуалізації та квантифікації розподілу хімічних речовин, наприклад сигнальних молекул, у клітинах.[79]
Флуоресцентний зонд складається з двох основних компонентів: 1) рецептору, що зв'язується з молекулою, яку треба визначити (в аналітичній хімії її називають аналітом); 2) флуорофору, що реагує на зміну оточення, змінюючи флуоресценцію.[17] Існує велика кількість механізмів, які здатні трансформувати зв'язування між рецептором та аналітом у зміну флуоресцентного сигналу. Наприклад, при зв'язуванні рецептору з аналітом може змінюватись конформація молекули, що призводить до подовження або скорочення системи спряжених π-зв'язків.[80] Зміна конформації молекули може впливати на відстань між FRET-парою, що також призведе до помітних змін у флуоресценції. Утворення нових координаційних зв'язків між рецептором та аналітом може активувати/блокувати перенесення електрону у збудженому стані (англ.: photoinduced electron transfer), який є одним із механізмів гасіння флуоресценції.[81] Існують також інші механізми.[82]
Флуоресцентний зонд може по-різному змінювати флуоресценцію при зв'язуванні з аналітом, що схематично показано на малюнку: флуоресценція може зростати (випадок А), спадати (В) або повністю змінювати один із параметрів, наприклад, колір (випадок С).
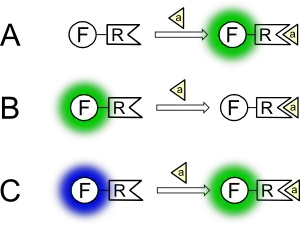
Прикладами для першого випадку (зростання флуоресценції в присутності аналіту) є численні похідні флуоресцеїну та родаміну у закритій лактонній формі. Розкриття лактонів з утворенням відкритої флуоресцентної форми при реакції з такими речовинами як перекис водню, сірководень або оксид азоту (NO) є методом виявлення цих біологічно-важливих молекул у живих організмах.[83] Прикладом для другого випадку (зменшення флуоресценції при взаємодії з аналітом) є флуоресцентні зонди на хлорид-іони: флуоресценція багатьох похідних хіноліну зменшується в присутності іонів хлору.[1] Нарешті, прикладом для третього випадком є Fura-2, один із перших ратіометричних зондів для іонів кальцію[84], який змінює колір флуоресценції при зміні концентрації іонів Ca2+ в середовищі.
У деяких випадках флуоресцентний зонд реагує не на присутність якоїсь окремої хімічної речовини, а зміну фізичних параметрів середовища, в якому він перебуває (температура, полярність, в'язкість). Важливим прикладом є сольватохромні флуоресцентні барвники — сполуки що змінюють колір флуоресценції залежно від полярності оточення. Сольватохромні флуоресцентні барвники стали важливим інструментом дослідження ліпідного складу та фазових переходів у ліпідних мембранах клітин.[85] Інша група сполук, яку називають флуоресцентними молекулярними роторами, змінює інтенсивність флуоресценції залежно від в'язкості середовища. Інтенсивність флуоресценції дуже низька у звичайних розчинниках, тоді як за високих значень динамічної в'язкості середовища інтенсивність флуоресценції зростає в десятки разів.[86] За допомогою флуоресцентних молекулярних роторів та конфокальної флуоресцентної мікроскопії стало можливим досліджувати в'язкість середовища всередині живих клітин.[87] Протягом останніх років флуоресцнтні зонди стали незамінними засобами дослідження живих клітин, збагативши клітинну біологію новими швидкими та точними методами кількісного аналізу.[88][89]
Приклади використання флуоресценції
Секвенування ДНК
Секвенуванням називають визначення послідовності нуклеотидів у ланцюгу нуклеїнової кислоти.[90] Перші методи секвенування були розроблені в 1970-х роках XX сторіччя. Ними були метод хімічної деградації Максама-Гілберта[91] та метод, що базується на використанні дідеокситермінаторів за Сангером.[92] Суть останнього полягала в ензиматичному подовженні праймеру (короткого олігонуклеотида-затравки відомої структури) на молекулі ДНК невідомої послідовності у присутності спеціальних хімічно-модифікованих нуклеозидів, що мають властивості припиняти ПЛР — дідеокситермінаторів. Вони схожі на звичайні нуклеозид трифосфати, які є вихідними сполуками для синтезу ДНК в організмі, але відрізняються від них відсутністю 3'-гідроксильної групи. Ці хімічні сполуки можуть інкорпоруватись в послідовність ДНК, що синтезується ДНК-полімеразою. Але після інкорпорації дідеокситермінатора синтез обривається через відсутність вільного 3'-гідроксилу для утворення нового фосфодіестерного зв'язку з наступним нуклеозидом.
В оригінальному методі Сангера використовувалось ензиматичне подовження праймеру, міченого радіоактивним ізотопом (32Р на 5' гідроксильній групі) в чотирьох різних пробірках. До кожної з них додавався невеликий відсоток одного певного дідеокситермінатора. Через це синтез в кожній пробірці обривався в певний момент, але завжди на позиції того нуклеотиду, який був у формі дідеокситермінатору. За теорією ймовірності, у суміші, що містить велику кількість новоутворених молекул ДНК, накопичувались всі можливі послідовності, терміновані на всіх можливих позиціях, що містять нуклеотид, присутній у формі ddNTP. Після завершення реакції всі чотири реакційні суміші розділялись в поліакриламідному гелі на чотирьох різних доріжках, розподілення радіоактивних фрагментів зчитувалось радіоавтографією, після чого послідовність невідомої ДНК можна було прочитати прямо на знімку.
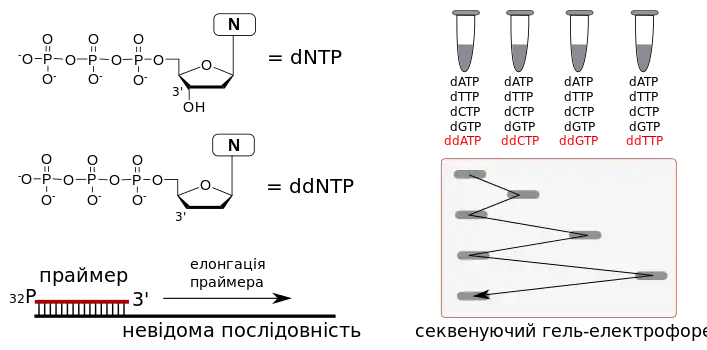
Окрім використання радіоактивних ізотопів, іншим недоліком цього методу була велика кількість операцій, необхідна для виявлення та зчитування радіоактивного сигналу, а також необхідність використання чотирьох доріжок для кожного аналізу, тому що неможливо було розрізнити різні терміновані фрагменти тільки за їх радіоактивністю.
Метод секвенування з дідеокситермінаторами був суттєво вдосконалений, коли радіоактивне мічення праймеру було замінено на флуоресцентне мічення термінальних нуклеотидів. На малюнку показана структура дідезоксинуклеозид-трифосфатів, які містять флуоресцентні барвники, прив'язані ковалентними зв'язками до азотистих основ. Було знайдено, що такі модифікації азотистих основ мінімально впливають на розпізнавання трифосфатів ДНК-полімеразами, тому вони можуть вбудовуватись у синтезовану ДНК наряд зі звичайними дНТФ. У випадку з флуоресцентними дідеокситермінаторами при термінації синтезу ДНК відбувається її флуоресцентне мічення. Використання флуоресцентних барвників чотирьох кольорів для кодування кожного з природних нуклеозидів дозволило проводити синтез в одній пробірці та розділення на одній доріжці гелю. Більш того, флуоресцентна детекція виявилась більш чутливою та швидкою за радіоактивну, дозволяючи проводити визначення нуклеотидів у реальному часі.
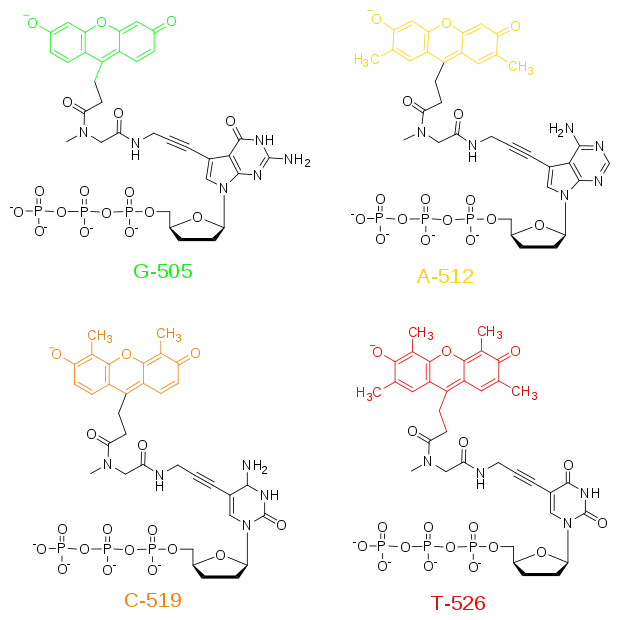
У результаті наприкінці 1980-х вдалося розробити автоматичні системи для секвенування ДНК із розділенням термінованих фрагментів у капілярному варіанті гель-електрофорезу та з детекцією кожної «букви» в послідовності за її специфічним кольором флуоресценції.
Хоча саме завдяки цьому методу була розшифрована значна частина ДНК людини, секвенування за Сангером вже не актуальне через існування більш швидких, дешевих та ефективних методів нових поколінь. Багато з них також базується на флуоресцентній детекції. Наприклад, секвенування за допомогою синтезу (sequencing by synthesis) також використовує кодування чотирма різними кольорами флуоресценції для кожної з чотирьох літер генетичного коду.[93]
Гібридизація ДНК
Молекули ДНК складаються з двох ланцюгів полінуклеотидів, які комплементарні одне одному. Азотисті основи двох ланцюгів утворюють пари, які стабілізовані водневими зв'язками. Характерною рисою нуклеїнових кислот є здатність до молекулярного впізнавання, завдяки якій одноланцюгові фрагменти ДНК мають спорідненість до комплементарних фрагментів.
На основі явища гібридизації були створені методи аналізу послідовностей нуклеїнових кислот із використанням синтетичних флуоресцентно-мічених олігонуклеотидів.
Одним із них є флуоресцентна гібридизація in situ (fluorescent in situ hybridization, FISH), яка використовується для виявлення точної локалізації певних послідовностей ДНК на метафазних хромосомах[1]. У флуоресцентній гібридизації in situ використовують синтетичні олігонуклеотиди-зонди. Кожна послідовність—зонд ковалентно з'єднана з флуорофором певного кольору. Такі зонди вводяться в клітини, після чого залишаються на певний час, для того щоб відбулась гібридизація між зондами та комплементарними регіонами хромосомної ДНК. Зонди, які не гібридизувались, видаляються промиванням, після чого характерне забарвлення хромосом вивчається за допомогою флуоресцентної мікроскопії.
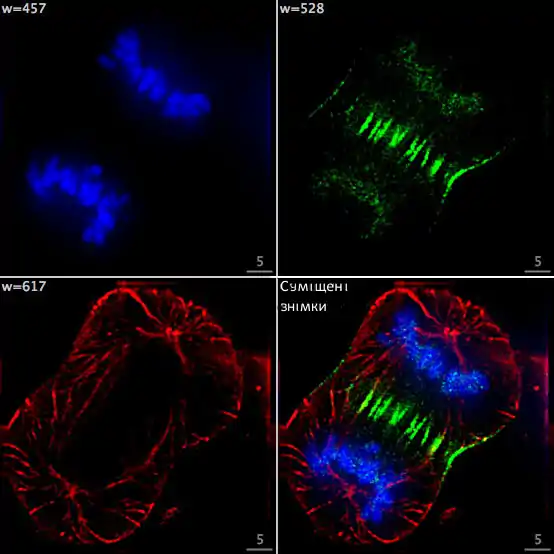
Окрім локалізації одиничних генів на хромосомах, FISH дозволяє досліджувати колокалізацію фрагментів ДНК. Завдяки цьому метод є корисним для цитології та генетики. Так, якщо використовувати для гібридизації з хромосомною ДНК два зонди, мічених червоним та зеленим флуорофорами, місця колокалізації цих послідовностей на хромосомах будуть виглядати як жовті точки.[1]
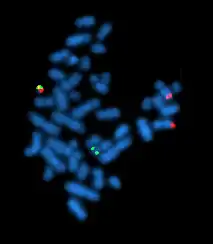
Часто стоїть задача визначити, чи міститься послідовність ДНК потрібної структури у розчині, наприклад у клітинному екстракті або у суміші продуктів полімеразної ланцюгової реакції. Флуоресцента гібридизація in situ для цього непридатна, тому що при зв'язувані міченої ДНК з мішенню не відбудеться зміни флуоресценції, а відділити зв'язаний та незв'язаний зонд як у випадку з хромосомами неможливо через їх однакову розчинність та інші фізико-хімічні властивості.
Елегантний метод для вирішення цієї задачі був знайдений в 1996-му році[94] та отримав назву molecular beacon probes (MB-зонди, буквальний переклад: «молекулярні маяки»). Структура та принцип роботи МВ-зондів зображені на малюнку.
МВ-зонд є одноланцюговим фрагментом ДНК, що складається з двох ділянок: петлі (червона) та основи (чорна). Послідовність нуклеотидів у петлі вибирається таким чином, щоб бути комплементарній тій послідовності, яку треба визначити (мішені). Дві частини основи комплементарні одна одній, і тому утворюють стабільну структуру за відсутності мішені. Кінцеві гідроксильні групи одноланцюгової ДНК ковалентно модифікуються флуорофором (F) з одного боку та гасником флуоресценції (Q) з іншого. За відсутності мішені зонд перебуває у закритому стані: флуорофор та гасник знаходяться одне біля одного, через що флуоресценція «вимкнена». У присутності ДНК-мішені може утворюватись гібридна структура в якій центральна петля комплементарна мішені, через що MB-зонд «розкривається». У такому стані кінці, що початково утворювали основу зонду, виявляються рознесені на значну відстань у просторі. Відповідно в такому стані гасник не може ефективно гасити флуорофор, що призводить до значного зростання інтенсивності флуоресценції.[94]
Оригінальний метод визначення ДНК за допомогою МВ-зондів зазнав чисельних вдосконалень[95]. Наприклад, існують модифікації, що базуються на використанні резонансного переносу енергії. Замість пари «флуорофор—гасник» кінці одноланцюгового ДНК-зонда мітять двома флуорофорами що утворюють FRET-пару; за рахунок цього наявність ДНК-мішені можна визначати за зникненням флуоресценції акцептора та зростанням флуоресценції донора.[96][97]
Важливою галуззю застосування МВ-зондів стала кількісна полімеразна ланцюгова реакція. МВ-зонд, комплементарний центральному регіону послідовності, що ампліфікується, додають у реакційну суміш до початку реакції. Після старту реакції інтенсивність флуоресценції вимірюється на кожному циклі ампліфікації. Якщо в результаті ПЛР ампліфікується фрагмент, комплементарний зонду, інтенсивність флуоресценції зростає пропорційно концентрації продукту в суміші. Завдяки цьому можливо оцінити кількість вихідної ДНК, що була на початку ампліфікації. Більш того, можливо спостерігати за ампліфікацією декількох варіантів послідовності ДНК в одній суміші, якщо використовувати комбінацію МВ-зондів закодованих різними кольорами флуоресценції. Цей підхід знайшов використання у генетичному аналізі для ідентифікації різних алелів одного гену.[98][99]
У флуоресцентній мікроскопії МВ — зонди використовуються для вивчення рівня експресії генів шляхом візуалізації мРНК в цитоплазмі. Коли певний ген починає транскриптуватися, у цитоплазмі зростає концентрація відповідної мРНК. Якщо синтезувати MB-зонд із петлею, що комплементарна ділянці мРНК, такий зонд буде гібридизуватись із нею, збільшуючи при цьому інтенсивність своєї флуоресценції. За рахунок цього можна дізнатись локалізацію відповідної мРНК в клітині та оцінити рівень експресії за зростанням флуоресценції.[100][101][102]
Мікромасиви ДНК
В організмах вищих еукаріотів містяться тисячі генів, сукупна робота яких визначає фенотип організму. Для швидкого одночасного дослідження великої кількості генів була розроблена технологія мікромасивів ДНК (англ. DNA microarrays).[103]
Мікромасив ДНК являє собою тверду поверхню, на яку нанесена велика кількість індивідуальних олігонуклеотидів. Кожен елемент масиву на поверхні містить ДНК однієї певної будови, яка програмується при створені масиву. Одним із методів створення ДНК мікромасивів є хімічний твердофазний синтез ДНК із використанням фотоактивних захисних груп.[104]
Ця технологія виявилась зручною для аналізу рівню експресії генів у клітинах. Для цього потрібен ДНК мікромасив, що містить набір олігонуклеотидних маркерів, специфічних для кожного з генів, які потрібно дослідити. Для того, щоб порівняти рівень експресії у двох зразках, контрольному і досліджуваному, проводять наступні операції.

Клітини обох зразків обробляють спеціальними хімічними реагентами та екстрагують матричну РНК, що міститься в цитоплазмі. РНК кожного зразку інкубують у присутності зворотньої траскриптази та флуоресцентних маркерів певного кольору, в результаті чого утворюються флуоресцентно-мічені молекули кДНК. Так, наприклад, на малюнку, що зображено вище, кДНК зразку А була помічена червоним флуороформ, а кДНК зразку В — зеленим. Після цього зразки змішують та гібридизують на мікромасиві. У результаті молекули кДНК гібридизуються в комірці, яка відповідає їхньому гену. Аналіз кольору флуоресценції в кожній комірці показує різницю в рівні експресії відповідного гену в обох зразках. Якщо комірка має червоний колір, це значить що при гібридизації розчину на мікромасиві в ньому містилось більше кДНК з клітин А, а отже рівень експресії гену в клітинах А був вищій. Якщо комірка має зелений колір, значить рівень експресії був вищим у клітинах В. Нарешті жовтий колір свідчить про те, що клітини містили однакову кількість мРНК, отже рівень експресії цього гену в них був однаковий.[103]
Флуоресцентна мікроскопія

Окремою галуззю застосування флуоресценції в біології є флуоресцентна мікроскопія — варіант оптичної мікроскопії, який базується на дослідженні флуоресцентних молекул у мікрооб'єктах.[17] Цей метод набув широкого розповсюдження та розквіту наприкінці XX-го століття. На відміну від традиційної оптичної мікроскопії, в якій контрастне зображення створюється завдяки різному поглинанню світла окремими частинами клітини, у флуоресцентній мікроскопії контрастне зображення створюється завдяки флуоресценції певних молекул у зразку. Завдяки особливостям конструкції флуоресцентних мікроскопів збудне світло не попадає в об'єктив, що дає змогу отримувати яскраве контрастне зображення на темному фоні. Використання фотомультиплікаторів як детектора робить метод дуже чутливим. Сучасні методи забарвлення зразків, такі як імунофлуоресцентне фарбування, або введення у клітину флуоресцентних маркерних білків, дає змогу забарвлювати окремі елементи клітини з точністю, як неможлива при використанні класичних технік забарвлення мікроскопічних зразків. Більш того, на базі флуоресцентній мікроскопії створені новітні методи побудови та обробки зображення, які значно перевершують за просторовим розділенням традиційну оптичну мікроскопію.
Низькомолекулярні органічні флуоресцентні барвники для клітинних органел
Деякі низькомолекулярні органічні барвники виявляють афінність до певних біомолекул або до цілих клітинних органел. Це явище використовується для селективного мультикольорового забарвлення клітин у флуоресцентній мікроскопії. Окремі приклади барвників наведені у таблиці.
| Назва | Структурна формула | Колір флуоресценції | Що забарвлює |
|---|---|---|---|
| DAPI 4',6-діамідино-2-феніліндол |
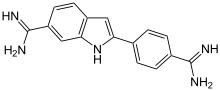 | синій | ядерна ДНК |
| Hoechst 33342 |
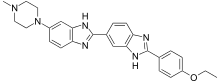 | синій | ядерна ДНК |
| Ніл червоний (Nile Red) |
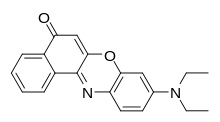 | червоний | ліпофільні елементи клітин (мембрани, ліпосоми) |
| MitoRed |
 | червоний | мітохондрії |
Одним із найрозповсюдженіших барвників для ядерної ДНК є 4',6-діамідино-2-феніліндол, DAPI. Він здатен селективно зв'язуватись із ДНК на А-Т збагачених ділянках, демонструючи яскраву синю флуоресценцію з максимумом на 461 нм.[105] Здатність забарвлювати хромосомну ДНК демонструють такі сполуки як Hoechst 33342, а також деякі ціанінові барвники.[106] Існують також флуоресцентні барвники, які переважно забарвлюють G-квадруплекси.[107]
Мітохондрії володіють великим негативним мембранним потенціалом (близько −180 mV), за рахунок чого можуть селективно забарвлюватись катіонними флуоресцентними барвниками, такими як MitoRed.[108]
Імунофлуоресцентне фарбування
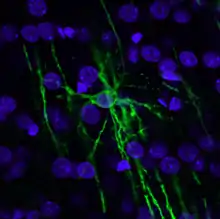
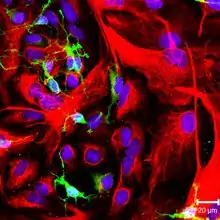
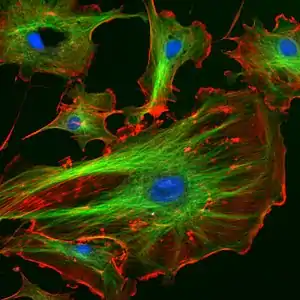
Суттєвим недоліком використання малих органічних сполук як флуоресцентних барвників є низька селективність мічення клітинних компонентів. Наприклад, сполуки, які зв'язуються з хромосомною ДНК можуть у тій чи іншій мірі забарвлювати інші нуклеїнові кислоти, що містяться в клітині; ліпофільні барвники, що забарвлюють ліпідні мембрани можуть також зв'язуватись із гідрофобними сайтами білків.
Селективного флуоресцентного мічення внутрішньоклітинних структур можна досягти за допомогою імунофлуоресцентного фарбування клітин. Цей метод поєднує селективність традиційних методів імунофарбування з чутливістю флуоресцентної детекції.
Як і класичне імунофарбування, цей метод базується на використанні антитіл. Антитіла — це білкові молекули, що з високою афінністю та селективністю зв'язуються зі своїми мішенями. Кожне антитіло розпізнає свій певний антиген.
В імунофлуоресцентному фарбуванні використовують одразу два типи антитіл. Первинне антитіло зв'язується безпосередньо з об'єктом фарбування. Після цього вторинне антитіло, яке ковалентно модифіковане молекулою флуорофору, зв'язується з первинним антитілом. Таким чином, мішень, що буде нести антиген забарвлюватиметься за рахунок утворення комплексу з двома антитілами.[17]
Використання двох антитіл, первинного і вторинного, необхідне для забезпечення гнучкості методу, тобто заради можливості варіювати забарвлення різних мішеней без необхідності отримання і хімічної модифікації нових антитіл.
Недоліком імунофлуоресцентного фарбування є низька проникність антитіл крізь клітинні мембрани. Внаслідок цього даний метод найчастіше використовується для фарбування фіксованих клітин.
Автофлуоресцентні білки
Генетично запрограмовані автофлуоресцентні білки, створені на базі зеленого флюоресцентного білку (GFP) та його аналогів, є незамінними флуоресцентними маркерами для клітинної та молекулярної біології.
Перевагами автофлуоресцентних білків є можливість їх візуалізації в клітині без введення будь-яких додаткових барвників або хімічних реагентів. Генетичне мічення клітинних білків флуоресцентними білковими маркерами можна реалізувати, не змінюючи конформацію та біохімічні властивості маркованого білка. Внаслідок чого маркування білків GFP (GFP fusion constructs) використовується для вивчення рівня експресії білків у клітинах.[109]
Окрім пасивних флуоресцентних маркерів, які просто сигналізують про наявність і локалізацію білка, до якого вони прив'язані, на базі GFP були створені флуоресцентні зонди, які змінюють флуоресценцію залежно від концентрації іонів, сигнальних молекул та інших факторів середовища, в якому вони знаходяться.[110]
Флуоресцентний мікроскоп
Для флуоресцентної мікроскопії необхідні мікроскопи спеціальної будови.[111] Сильно спрощена схема такого мікроскопу (а саме схема растрового конфокального епіфлуоресцентного мікроскопу) показана на малюнку нижче. Він має наступні особливості будови:[17][111]
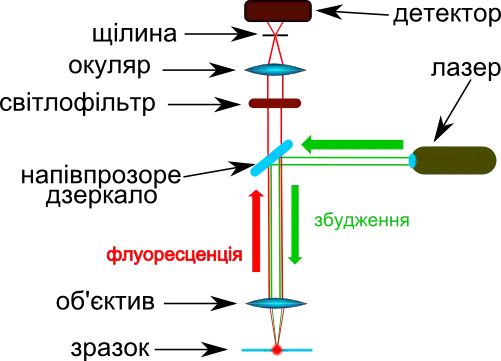
- Як джерела світла використовуються лазери, або комбінація звичайних джерел світла та монохроматорів. Це дає змогу селективно опромінювати зразок світлом певної довжини хвилі для збудження флуорофорів одного типу.
- Використання напівпрозорого дзеркала спрощує конструкцію, даючи змогу використовувати один і той самий оптичний шлях для збудного (зелені лінії на малюнку) та флуоресцентного (червоні лінії) світла. На відміну від класичної мікроскопії, в якій опромінюється весь зразок одночасно, такі епіфлуоресцентні мікроскопи направляють збудне світло тільки на ту ділянку препарату, яка безпосередньо знаходиться в полі зору. Всі інші частини зображення залишаються в темряві. Це дозволяє мінімізувати фотознебарвлення нестійких флуорофорів та зменшує фототоксичні ефекти.
- Якість зображень суттєво покращується при використанні конфокальних флуоресцентних мікроскопів. У таких мікроскопах на шляху флуоресцентного світла встановлюється щілина (англ. pinhole), через що вдається спостерігати флуоресценцію лише від тієї частини зразку, яка знаходиться у фокусі зображення. За допомогою конфокальних флуоресцентних мікроскопів можливо змінювати глибину проникнення в клітину, спостерігаючи за окремими її «зрізами» (фізично клітина залишається цілою). Це також дає змогу зробити тривимірну реконструкцію клітини, накопичивши достатню кількість оптичних «зрізів».
- Використання фотомультиплікатора як детектора дозволяє детектувати окремі фотони, що значно підвищує чутливість методу.
- У растровому флуоресцентному мікроскопі зображення створюється комп'ютером після сканування зразку. Світло лазеру почергово направляється в кожну точку зображення; при цьому програмне забезпечення мікроскопу записує інтенсивність флуоресценції та відносить її до координат точки, з якої вона була отримана. Після сканування всіх точок у полі зору, зображення реконструюється на екрані комп'ютера.
Окрім растрових конфокальних мікроскопів існують інші типи цих приладів.
Флуоресцентна мікроскопія надвисокої роздільної здатності

У растровому флуоресцентному конфокальному мікроскопі зображення створюється послідовним переміщенням променю лазеру від точки до точки кадру, реєстрацією флуоресценції та наступною реконструкцією зображення на комп'ютері. Як виявилось, цей метод має певні недоліки. Явище дифракції накладає певні фізичні обмеження на мінімальний розмір сфокусованого лазерного променю, а отже і на максимальне просторове розділення методу.
Згодом з'ясувалось, що цей дифракційний бар'єр може бути подоланий багатьма різними методами, що відкривало можливості для оптичної флуоресцентної мікроскопії надвисокої роздільної здатності.[112][113]
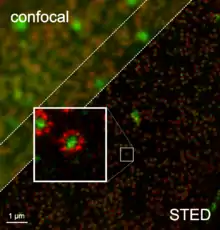
Одним із перших способів стала STED мікроскопія (від англ. stimulated emission depletion). В основі цього методу лежить взаємодія флуорофорів, збуджених звичайним лазерним імпульсом із наступним STED-імпульсом. Якщо збуджені молекули флуорофору опромінити електромагнітним імпульсом, який за енергією відповідає очікуваній енергії флуоресценції, відбувається примусове пригнічення емісії (stimulated emission depletion). Флуорофори, що потрапили під STED-імпульс, не можуть флуоресціювати. Якщо варіювати фази збудного та STED-імпульсів, можна отримати різну їх локалізацію у просторі. Так, малюнок справа показує проекцію початкового лазерного імпульсу, форму STED-імпульсу, та ділянку що містить збуджені флуорофори після накладання двох імпульсів.
Завдяки цьому було зменшено площу, з якої реєструється флуоресценція, а отже підвищено просторове розділення методу. На фотографії внизу показані для порівняння два зображення одного і того ж об'єкту, зроблені із використанням растрової конфокальної та STED-мікроскопії.
Примітки
- Joseph R. Lakowicz. Principles of Fluorescence Spectroscopy. — Springer Science+Business Media, 2006. — ISBN 978-0387-31278-1.
- Bernard Valeur. Molecular Fluorescence Principles and Applications. — Wiley-VCH Verlag GmbH. — ISBN 3-527-29919-X.
- Millar, David P. Fluorescence studies of DNA and RNA structure and dynamics // Current Opinion in Structural Biology. — 1996. — Т. 6. — С. 322-326. — DOI:.
- Royer, C. A. Probing Protein Folding and Conformational Transitions with Fluorescence // Chemical Reviews. — 2006. — Т. 106, вип. 5. — С. 1769-1784. — DOI:.
- Smith, L. M., Sanders, J. Z., Kaiser, R. J., Hughes, P., Dodd, C., Connell, C. R., Heiner, C., Kent, S. B. H., & Hood, L. E. (1986). Fluorescence detection in automated DNA sequence analysis. Nature, 321(6071), 674—679.
- Prober, J. M., Trainor, G. L., Dam, R. J., Hobbs, F. W., Robertson, C. W., Zagursky, R. J., Cocuzza, A. J., Jensen, M. A., & Baumeister, K. (1987). A system for rapid DNA sequencing with fluorescent chain-terminating dideoxynucleotides. Science, 238(4825), 336—341.
- Shendure, J., & Ji, H. (2008). Next-generation DNA sequencing. Nature Biotechnology, 26(10), 1135—1145.
- McGinn, S., & Gut, I. G. (2012). DNA sequencing — spanning the generations. New Biotechnology.
- Schepartz, A.; Gonzalez, R. L. Molecular imaging: sine labore nihil // Current Opinion in Chemical Biology. — 2011. — Т. 15. — С. 749–751. — DOI:.
- Drummen, G. P. C. Fluorescent Probes and Fluorescence (Microscopy) Techniques — Illuminating Biological and Biomedical Research // Molecules. — 2012. — Т. 17. — С. 14067-14090. — DOI:.
- O'Haver, T. C. Development of luminescence spectrometry as an analytical tool // Journal of Chemical Education. — 1978. — Т. 55, вип. 7. — С. 423—428. — DOI:.
- Rao, J.; Dragulescu-Andrasi, A.; Yao, H. Fluorescence imaging in vivo: recent advances // Current Opinion in Biotechnology. — 2007. — Т. 18. — С. 17–25. — DOI:.
- Hilderbrand, S. A.; Weissleder, R. Near-infrared fluorescence: application to in vivo molecular imaging // Current Opinion in Chemical Biology. — 2010. — Т. 14. — С. 71–79. — DOI:.
- Kobayashi, H., Ogawa, M., Alford, R., Choyke, P. L., & Urano, Y. New Strategies for Fluorescent Probe Design in Medical Diagnostic Imaging // Chemical Reviews. — 2010. — Т. 110, вип. 5. — С. 2620-2640. — DOI:.
- Gioux, S., Choi, H. S., & Frangioni, J. V. Image-Guided Surgery using Invisible Near-Infrared Light: Fundamentals of Clinical Translation // Molecular Imaging. — 2010. — Т. 9, вип. 5. — С. 237–255.
- Alander, J. T., Kaartinen, I., Laakso, A., Patila, T., Spillmann, T., Tuchin, V. V., Venermo, M., Valisuo, P. A Review of Indocyanine Green Fluorescent Imaging in Surgery. // International Journal of Biomedical Imaging. — 2012. — DOI:. — 940585.
- Alexander P. Demchenko. Introduction to Fluorescence Sensing. — Springer Science + Business Media B.V, 2009. — ISBN 978-1-4020-9002-8.
- Udenfriend, S. Development of the spectrophotofluorometer and its commercialization // Protein Science. — 1995. — Т. 4, вип. 3. — С. 542-551. — DOI:.
- Е. В. КУДРЯШОВА, А. К. ГЛАДИЛИН, А. В. ЛЕВАШОВ. БЕЛКИ В НАДМОЛЕКУЛЯРНЫХ АНСАМБЛЯХ: ИССЛЕДОВАНИЕ СТРУКТУРЫ МЕТОДОМ РАЗРЕШЕННО–ВРЕМЕННОЙ ФЛУОРЕСЦЕНТНОЙ АНИЗОТРОПИИ // Успехи биологической химии. — 2002. — Т. 42 (21 січня). — С. 257-294.
- Weiss, S. Measuring conformational dynamics of biomolecules by single molecule fluorescence spectroscopy // Nature Structural Biology. — 2000. — Т. 7, вип. 9. — С. 724—729. — DOI:.
- Selvin, P. R. The renaissance of fluorescence resonance energy transfer // Nature Structural Biology. — 2000. — Т. 7, вип. 9. — С. 730-734. — DOI:.
- Klostermeier, D., Millar, D. P. Time-resolved fluorescence resonance energy transfer: A versatile tool for the analysis of nucleic acids // Biopolymers. — 2002. — Т. 61, вип. 3. — С. 159–179. — DOI:.
- Ambrose, W. P., Goodwin, P. M., Jett, J. H., Van Orden, A., Werner, J. H., & Keller, R. A. Single Molecule Fluorescence Spectroscopy at Ambient Temperature // Chemical Reviews. — 1999. — Т. 99, вип. 10. — С. 2929-2956. — DOI:.
- Tinnefeld, P., & Sauer, M. Branching Out of Single-Molecule Fluorescence Spectroscopy: Challenges for Chemistry and Influence on Biology. // Angewandte Chemie International Edition. — 2005. — Т. 44. — С. 2642–2671. — DOI:.
- Ha, T. Single-molecule fluorescence methods for the study of nucleic acids // Current Opinion in Structural Biology. — 2001. — Т. 11. — С. 287–292. — DOI:.
- Haran, G. Single-molecule fluorescence spectroscopy of biomolecular folding // Journal of Physics: Condensed Matter. — 2003. — Т. 15. — С. 1292-1313. — DOI:.
- Michalet, X., Weiss, S., & Jäger, M. Single-Molecule Fluorescence Studies of Protein Folding and Conformational Dynamics // Chemical Reviews. — 2006. — Т. 106, вип. 5. — С. 1785-1813. — DOI:.
- Hohlbein, J., Gryte, K., Heilemann, M., & Kapanidis, A. N. Surfing on a new wave of single-molecule fluorescence methods // Physical Biology. — 2010. — Т. 7, вип. 3. — С. 031001. — DOI:.
- Moerner, W. E.; Fromm, D. P. Methods of single-molecule fluorescence spectroscopy and microscopy // Review of Scientific Instruments. — 2003. — Т. 74. — С. 3597—3619. — DOI:.
- Persson, F., Barkefors, I., & Elf, J. Single molecule methods with applications in living cells // Current Opinion in Biotechnology. — 2013. — Т. 24, вип. 4. — С. 737–744. — DOI:.
- Delehanty, J. B., Bradburne, C. E., Susumu, K., Boeneman, K., Mei, B. C., Farrell, D., Blanco-Canosa, J. B., Dawson, P. E., Mattoussi, H., & Medintz, I. L. Spatiotemporal Multicolor Labeling of Individual Cells Using Peptide-Functionalized Quantum Dots and Mixed Delivery Techniques // Journal of the American Chemical Society. — 2011. — Т. 133, вип. 27. — С. 10482-10489. — DOI:.
- Ko, S.-K.; Chen, X.; Yoon, J.; Shin, I. Zebrafish as a good vertebrate model for molecular imaging using fluorescent probes // Chemical Society Reviews. — 2011. — Т. 40, вип. 5. — С. 2120-2130. — DOI:.
- Bo Huang. Super-resolution optical microscopy: multiple choices // Current Opinion in Chemical Biology. — 2010. — Т. 14, вип. 1. — С. 10–14. — DOI:.
- Lothar Schermelleh, Rainer Heintzmann, Heinrich Leonhardt. A guide to super-resolution fluorescence microscopy // The Journal of Cell Biology. — 2010. — Т. 190, вип. 2. — С. 165. — DOI:.
- Alexander P. Demchenko. Ultraviolet Spectroscopy of Proteins. — Springer, 1986. — ISBN 978-3642708497.
- Crespo-Hernandez, C. E.; Cohen, B.; Hare, P. M.; Kohler, B. Ultrafast Excited-State Dynamics in Nucleic Acids // Chemical Reviews. — 2004. — Т. 104, вип. 4. — С. 1977-2020. — DOI:.
- Waggoner, A. Fluorescent labels for proteomics and genomics // Current Opinion in Chemical Biology. — 2006. — Т. 10, вип. 1. — С. 62—66. — DOI:.
- Du, W.; Wang, Y.; Luo, Q.; Liu, B.-F. Optical molecular imaging for systems biology: from molecule to organism // Analytical and Bioanalytical Chemistry. — 2006. — Т. 386, вип. 3. — С. 444—457. — DOI:.
- Lavis, L. D. Histochemistry: Live and in Color // Journal of Histochemistry & Cytochemistry. — 2011. — Т. 59, вип. 2. — С. 139—145. — DOI:.
- Ueno, T.; Nagano, T. Fluorescent probes for sensing and imaging // Nature Methods. — 2011. — Т. 8. — С. 642-645. — DOI:.
- Lavis, L. D.; Raines, R. T. Bright Ideas for Chemical Biology // ACS Chemical Biology. — 2008. — Т. 3, вип. 3. — С. 142-155. — DOI:.
- Zheng, H.; Zhan, X.-Q.; Bian, Q.-N.; Zhang, X.-J. Advances in modifying fluorescein and rhodamine fluorophores as fluorescent chemosensors // Chemical Communications. — 2012. — Т. 49. — С. 429-447. — DOI:.
- Beija, M.; Afonso, C. A. M.; Martinho, J. M. G. Synthesis and applications of Rhodamine derivatives as fluorescent probes // Chemical Society Reviews. — 2009. — Т. 38, вип. 8. — С. 2410-2433. — DOI:.
- Boens, N.; Leen, V.; Dehaen, W. Fluorescent indicators based on BODIPY // Chemical Society Reviews. — 2012. — Т. 41. — С. 1130–1172.
- Tatikolov, A. S. Polymethine dyes as spectral-fluorescent probes for biomacromolecules // Journal of Photochemistry and Photobiology C: Photochemistry Reviews. — 2012. — Т. 13, вип. 1. — С. 55–90. — DOI:.
- Beverina, L.; Salice, P. Squaraine Compounds: Tailored Design and Synthesis towards a Variety of Material Science Applications // European Journal of Organic Chemistry. — 2010. — С. 1207-1225. — DOI:.
- Chen, X.; Pradhan, T.; Wang, F.; Kim, J. S.; Yoon, J. Fluorescent Chemosensors Based on Spiroring-Opening of Xanthenes and Related Derivatives // Chemical Reviews. — 2012. — Т. 112. — С. 1910–1956. — DOI:.
- Yuan, L.; Lin, W.; Zheng, K.; He, L.; Huang, W. Far-red to near infrared analyte-responsive fluorescent probes based on organic fluorophore platforms for fluorescence imaging // Chemical Society Reviews. — 2013. — Т. 42. — С. 622-661. — DOI:.
- Grimm, J. B.; Heckman, L. M.; Lavis, L. D.; May, C. M. The Chemistry of Small-Molecule Fluorogenic Probes // Progress in Molecular Biology and Translational Science. — 2013. — Т. 113. — С. 1-13. — DOI:.
- Wysocki, L. M.; Lavis, L. D. Advances in the chemistry of small molecule fluorescent probes // Current Opinion in Chemical Biology. — 2011. — Т. 15, вип. 6. — С. 752—759. — DOI:.
- Sun, Y.-Q.; Liu, J.; Lv, X.; Liu, Y.; Zhao, Y.; Guo, W. Rhodamine-Inspired Far-Red to Near-Infrared Dyes and Their Application as Fluorescence Probes // Angewandte Chemie International Edition. — 2012. — Т. 51, вип. 31. — С. 7634–7636. — DOI:.
- Finney, N. S. Combinatorial discovery of fluorophores and fluorescent probes // Current Opinion in Chemical Biology. — 2006. — Т. 10, вип. 3. — С. 238-245. — DOI:.
- Bünzli, J.-C. G. Lanthanide Luminescence for Biomedical Analyses and Imaging // Chemical Reviews. — Т. 110. — С. 2729–2755. — DOI:.
- Shimomura O, Johnson F, Saiga Y (1962). «Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea». J Cell Comp Physiol 59 (3): 223-39. doi:10.1002/jcp.1030590302.
- Ormö M, Cubitt A, Kallio K, Gross L, Tsien R, Remington S. Crystal structure of the Aequorea victoria green fluorescent protein // Science. — 1996. — Т. 273. — С. 1392–1395. — DOI:.
- Yang F, Moss L, Phillips G. The molecular structure of green fluorescent protein // Nature Biotechnology. — 1996. — Т. 14, вип. 10. — С. 1246–1251. — DOI:.
- Tsien, R. Y. The Green Fluorescent Protein // Annual Review of Biochemistry. — 1998. — Т. 67, вип. 1. — С. 509-544. — DOI:.
- Zimmer, M. Green Fluorescent Protein (GFP): Applications, Structure, and Related Photophysical Behavior // Chemical Reviews. — 2002. — Т. 102, вип. 3. — С. 759-782. — DOI:.
- Beat Ludin, Andrew Matus. GFP illuminates the cytoskeleton // Trends in Cell Biology. — 1998. — Т. 8, вип. 2. — С. 72–77. — DOI:.
- Tsien, R. Y. Building and breeding molecules to spy on cells and tumors // FEBS Letters. — 2005. — Т. 579, вип. 4. — С. 927-932. — DOI:.
- Giepmans, B. N. G., Adams, S. R., Ellisman, M. H., & Tsien, R. Y. (2006). The Fluorescent Toolbox for Assessing Protein Location and Function. Science, 312(5771), 217—224.
- Stepanenko, O. V.; Stepanenko, O. V.; Shcherbakova, D. M.; Kuznetsova, I. M.; Turoverov, К. К.; Verkhusha, V. V. Modern f luorescent proteins: from chromophore formation to novel intracellular applications // BioTechniques. — 2011. — Т. 51. — С. 313—327. — DOI:.
- Zhou, X. X.; Lin, M. Z. Photoswitchable fluorescent proteins: ten years of colorful chemistry and exciting applications // Current Opinion in Chemical Biology. — 2013. — Т. 17, вип. 4. — С. 682-690. — DOI:.
- Ohba, Y., Fujioka, Y., Nakada, S., Tsuda, M., & May, C. M. Fluorescent Protein-Based Biosensors and Their Clinical Applications. // Progress in Molecular Biology and Translational Science. — 2013. — Т. 113. — С. 313-348. — DOI:.
- VanEngelenburg, S. B.; Palmer, A. E.,. Fluorescent biosensors of protein function // Current Opinion in Chemical Biology. — 2008. — Т. 12. — С. 60–65. — DOI:.
- Tiwari, D. K.; Nagai, T. Smart fluorescent proteins: Innovation for barrier-free superresolution imaging in living cells // Development, Growth & Differentiation. — 2013. — Т. 55, вип. 4. — С. 491-507. — DOI:.
- The Nobel Prize in Chemistry 2008
- Jyoti K. Jaiswal, Sanford M. Simon. Potentials and pitfalls of fluorescent quantum dots for biological imaging // Trends in Cell Biology. — 2004. — Т. 14, вип. 9 (21 січня). — С. 497–504. — DOI:.
- Biju, V., Itoh, T., & Ishikawa, M. (2010). Delivering quantum dots to cells: bioconjugated quantum dots for targeted and nonspecific extracellular and intracellular imaging. Chemical Society Reviews, 39(8), 3031-3056.
- Jin, Z., & Hildebrandt, N. (2012). Semiconductor quantum dots for in vitro diagnostics and cellular imaging. Trends in Biotechnology.
- Vivero-Escoto, J. L., Huxford-Phillips, R. C., & Lin, W. Silica-based nanoprobes for biomedical imaging and theranostic applications. // Chem. Soc. Rev.. — 2012. — Т. 41 (21 січня). — С. 2673–2685. — DOI:.
- Wu, C.; Chiu, D. T. Highly Fluorescent Semiconducting Polymer Dots for Biology and Medicine // Angewandte Chemie International Edition. — 2013. — Т. 52 (21 січня). — С. 3086-3109. — DOI:.
- Han, B., & Wang, E. DNA-templated fluorescent silver nanoclusters // Analytical and Bioanalytical Chemistry. — 2012. — Т. 402, вип. 1 (21 січня). — С. 129-138. — DOI:.
- Shiang, Y.-C.; Huang, C.-C.; Chen, W.-Y.; Chen, P.-C.; Chang, H.-T. Fluorescent gold and silver nanoclusters for the analysis of biopolymers and cell imaging // Journal of Materials Chemistry. — 2012. — Т. 22. — С. 12972–12982. — DOI:.
- Lemke, E. A.; Schultz, C. Principles for designing fluorescent sensors and reporters // Nature Chemical Biology. — 2011. — Т. 7. — С. 480–483. — DOI:.
- J. C. Simpson, B. Joggerst, V. Laketa, F. Verissimo, C. Cetin, H. Erfle, M. G. Bexiga, V. R. Singan, J.-K. Hériché, B. Neumann, A. Mateos, J. Blake, S. Bechtel, V. Benes, S. Wiemann, J. Ellenberg, R. Pepperkok. Genome-wide RNAi screening identifies human proteins with a regulatory function in the early secretory pathway // Nature Cell Biology. — 2012. — Т. 14 (21 січня). — С. 764–774. — DOI:.
- Susan M. Gasser. Visualizing Chromatin Dynamics in Interphase Nuclei // Science. — 2002. — Т. 2002, вип. 296 (21 січня). — С. 1412-1416. — DOI:.
- Nathalie Daigle, Jan Ellenberg. λN-GFP: an RNA reporter system for live-cell imaging // Nature Methods. — 2007. — Т. 4, вип. 8 (21 січня). — С. 633-636. — DOI:.
- Johnson, I. Fluorescent probes for living cells // The Histochemical Journal. — 1998. — Т. 30, вип. 3. — С. 123-140. — DOI:.
- Shi, W.; Ma, H. Spectroscopic probes with changeable π-conjugated systems // Chemical Communications. — 2012. — Т. 48. — С. 8732-8744. — DOI:.
- Liu, Z.; He, W.; Guo, Z. Metal coordination in photoluminescent sensing // Chemical Society Reviews. — 2013. — Т. 42, вип. 4. — С. 1568-1600. — DOI:.
- de Silva, A. P., Gunaratne, H. Q. N., Gunnlaugsson, T., Huxley, A. J. M., McCoy, C. P., Rademacher, J. T., & Rice, T. E. Signaling Recognition Events with Fluorescent Sensors and Switches // Chemical Reviews. — 1997. — Т. 97, вип. 5. — С. 1515-1566. — DOI:.
- Chan, J.; Dodani, S. C.; Chang, C. J. Reaction-based small-molecule fuorescent probes for chemoselective bioimaging // Nature Chemistry. — 2012. — Т. 4. — С. 973-984. — DOI:.
- Grynkiewicz, G.; Poenie, M.; Tsien, R. Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties // Journal of Biological Chemistry. — 1985. — Т. 260. — С. 3440-3450.
- Demchenko, A. P.; Mély, Y.; Duportail, G.; Klymchenko, A. S. Monitoring Biophysical Properties of Lipid Membranes by Environment-Sensitive Fluorescent Probes // Biophysical Journal. — 2009. — Т. 96, вип. 9. — С. 3461-3470. — DOI:.
- Haidekker, M. A.; Theodorakis, E. A. Molecular rotors—fluorescent biosensors for viscosity and flow // Organic & Biomolecular Chemistry. — 2007. — Т. 5. — С. 1669–1678. — DOI:.
- Kuimova, M. K. Mapping viscosity in cells using molecular rotors // Physical Chemistry Chemical Physics. — 2012. — Т. 14. — С. 12671–12686. — DOI:.
- Johnsson, N.; Johnsson, K. Chemical Tools for Biomolecular Imaging // ACS Chemical Biology. — 2007. — Т. 2, вип. 1. — С. 31-38. — DOI:.
- Terai, T.; Nagano, T. Fluorescent probes for bioimaging applications // Current Opinion in Chemical Biology. — 2008. — Т. 12, вип. 515–521. — DOI:.
- Franca, L. T. C., Carrilho, E., & Kist, T. B. L. A review of DNA sequencing techniques // Quarterly Reviews of Biophysics. — 2002. — Т. 35, вип. 02. — С. 169-200. — DOI:.
- Maxam, A. M.; Gilbert, W. A new method for sequencing DNA // Proceedings of the National Academy of Sciences. — 1977. — Т. 74, вип. 2. — С. 560-564.
- Sanger, F.; Nicklen, S.; Coulson, A. R. DNA sequencing with chain-terminating inhibitors // Proceedings of the National Academy of Sciences. — 1977. — Т. 74, вип. 12. — С. 5463-5467.
- Ju, J.; Kim, D. H.; Bi, L.; Meng, Q.; Bai, X.; Li, Z.; Li, X.; Marma, M. S.; Shi, S.; Wu, J.; Edwards, J. R.; Romu, A.; Turro, N. J. Four-color DNA sequencing by synthesis using cleavable fluorescent nucleotide reversible terminators // Proceedings of the National Academy of Sciences. — 2006. — Т. 103, вип. 52. — С. 19635-19640. — DOI:.
- Tyagi, S.; Kramer, F. R. Molecular Beacons: Probes that Fluoresce upon Hybridization // Nature Biotechnology. — Т. 14, вип. 3. — С. 303-308. — DOI:.
- Guo, J.; Ju, J.; Turro, N.,. Fluorescent hybridization probes for nucleic acid detection. // Analytical and Bioanalytical Chemistry. — 2011. — Т. 402, вип. 10. — С. 3115-3125. — DOI:.
- Didenko, V. V. DNA Probes Using Fluorescence Resonance Energy Transfer (FRET): Designs and Applications (pdf) // BioTechniques. — 2001. — Т. 31. — С. 1106-1121.
- Juskowiak, B. Nucleic acid-based fluorescent probes and their analytical potential // Analytical and Bioanalytical Chemistry. — Т. 399, вип. 9. — С. 3157-3176. — DOI:.
- Tyagi S, Bratu DP, Kramer FR. Multicolor molecular beacons for allele discrimination // Nature Biotechnology. — 1998. — Т. 16. — С. 49—53. — DOI:.
- Ranasinghe, R. T.; Brown, T. Fluorescence based strategies for genetic analysis // Chemical Communications. — 2005. — С. 5487–5502. — DOI:.
- Silverman, A. P.; Kool, E. T. Quenched probes for highly specific detection of cellular RNAs // Trends in Biotechnology. — 2005. — Т. 23, вип. 5. — С. 225-230. — DOI:.
- Tyagi, S. Imaging intracellular RNA distribution and dynamics in living cells // Nature Methods. — 2009. — Т. 6, вип. 5. — С. 331-338. — DOI:.
- Wang, K.; Huang, J.; Yang, X.; He, X.; Liu, J. Recent advances in fluorescent nucleic acid probes for living cell studies // Analyst. — 2013. — Т. 138. — С. 62–71. — DOI:.
- Sassolas, A.; Leca-Bouvier, B. D.; Blum, L. J. DNA Biosensors and Microarrays // Chemical Reviews. — 2008. — Т. 108. — С. 109−139. — DOI:.
- Pirrung, M. C. Spatially Addressable Combinatorial Libraries // Chemical Reviews. — 1997. — Т. 97, вип. 2. — С. 473-488. — DOI:.
- Kapuscinski, J. DAPI: a DNA-Specific Fluorescent Probe // Biotechnic & Histochemistry. — 1995. — Т. 70, вип. 5. — С. 220-233. — DOI:.
- Yarmoluk, S. M.; Kovalska, V. B.; Losytskyy, M. Y. Symmetric cyanine dyes for detecting nucleic acids // Biotechnic & Histochemistry. — 2008. — Т. 83. — С. 131-145. — DOI:.
- Vummidi, B. R.; Alzeer, J.; Luedtke, N. W. Fluorescent Probes for G-Quadruplex Structures // ChemBioChem. — 2013. — Т. 14, вип. 5. — С. 540-558. — DOI:.
- Neto, B. A. D.; Correa, J. R.; Silva, R. G.,. Selective mitochondrial staining with small fluorescent probes: importance, design, synthesis, challenges and trends for new markers. // RSC Advances. — 2013. — Т. 3. — С. 5291-5301. — DOI:.
- Wahlfors, J.; Loimas, S.; Pasanen, T.; Hakkarainen, T. Green fluorescent protein (GFP) fusion constructs in gene therapy research // Histochemistry and Cell Biology. — 2001. — Т. 115. — С. 59-65. — DOI:.
- Zhang, J.; Campbell, R. E.; Ting, A. Y.; Tsien, R. Y. Creating new fluorescent probes for cell biology // Nature Reviews Molecular Cell Biology. — 2002. — Т. 3, вип. 12. — С. 906-918. — DOI:.
- Lichtman, J. W.; Conchello, J.-A. Fluorescence microscopy // Nature Methods. — 2005. — Т. 2, вип. 12. — С. 910-919. — DOI:.
- Bo Huang, Hazen Babcock, Xiaowei Zhuang. Breaking the Diffraction Barrier: Super-Resolution Imaging of Cells // Cell. — 2010. — Т. 143. — С. 1047-1058. — DOI:.
- Hell, S. W. Far-Field Optical Nanoscopy // Science. — 2007. — Т. 316. — С. 1153-1158. — DOI:.
Бібліографія
- Joseph R. Lakowicz. Principles of Fluorescence Spectroscopy. — Springer, 2006.
- Alexander P. Demchenko. Introduction to Fluorescence Sensing. — Springer Science + Business Media B.V, 2009. — ISBN 978-1-4020-9002-8.
|
|
Ця стаття належить до вибраних статей Української Вікіпедії. |