Програмована клітинна загибель
Програмована клітинна загибель, або програмована клітинна смерть[1], або запрограмована загибель клітин[2] (ПКЗ, англ. Programmed cell death) — загибель клітини, яка відбувається за рахунок запрограмованих внутрішньоклітинних процесів. До другого десятиліття XXI століття налічується більше десяти відомих видів програмованої клітинної загибелі. З 2005 року класифікацією видів клітинної загибелі займається Комітет з номенклатури видів клітинної загибелі (англ. Nomenclature Commitee on Cell Death). Програмована клітинна загибель описана для всіх великих груп еукаріотів: тварин, рослин, грибів, слизовиків і навіть одноклітинних організмів (наприклад, дріжджів). ПКЗ виконує багато функцій як на рівні клітини, так і на рівні цілого організму: у тварин вона відіграє важливу роль у розвитку, з її допомогою елімінуються пошкоджені клітини, у рослин вона задіяна в утворенні тканин, що складаються з мертвих клітин, таких як ксилема. Програмована клітинна загибель відома не тільки в еукаріотів: кілька видів програмованої загибелі було описано у бактерій[3]. Всі види програмованої клітинної загибелі можна поділити на зовнішні, які запускаються сигналами ззовні клітини, і внутрішні, викликані порушеннями у функціюванні клітин[4].
Класифікація
З точки зору морфології довгий час виділялося три основних види програмованої клітинної загибелі:
- Клітинна загибель I типу, або апоптоз. При цій формі клітинної загибелі відбуваються стиснення цитоплазми, конденсація хроматину, фрагментація ядра і так званий блебінг клітинної мембрани, тобто відбруньковування від неї везикул. Зрештою весь вміст клітини розпадається на везикули (апоптотичні тільця), які фагоцитуються сусідніми клітинами і розщеплюються в їхніх лізосомах.
- Клітинна загибель II типу, або автофагія. При автофагії в цитоплазмі підупалої клітини формується безліч вакуолей, які потім фагоцитуються і руйнуються сусідніми клітинами.
- Клітинна загибель III типу, або некроз. Некроз характеризується повною відсутністю рис, притаманних апоптозу та автофагії. Залишки зруйнованої клітини запускають запалення[4].
Пізніше було прийнято більш складну класифікацію видів програмованої клітинної загибелі, яка побудована не на морфологічних деталях, а на генетичних, біохімічних, фармакологічних і функціональних особливостях. Однак виділені таким чином види загибелі далі ділять на дві групи за морфологією: до однієї відносять види смерті, які морфологічно близькі до апоптозу, а в іншу — ті, які морфологічно близькі до некрозу. Таким чином, для кожного виду програмованої клітинної загибелі властивий свій набір властивостей, від повністю апоптотичного до повністю некротичного[4].
Станом на 2018 рік виділяють такі види програмованої клітинної загибелі[4]:
- Залежна від лізосом клітинна загибель (англ. Lysosome-dependent cell death, LDCD);
- Залежна від автофагії клітинна загибель (англ. Autophagy-dependent cell death, ADCD);
- Імуногенна клітинна загибель (англ. Immunogenic cell death, ICD);
- Внутрішній апоптоз (англ. Intrinsic apoptosis);
- Зовнішній апоптоз (англ. Extrinsic apoptosis);
- Некроз, залежний від проникності мітохондрій (MPT) (англ. MPT-driven necrosis);
- Некроптоз (англ. Necroptosis);
- Фероптоз (англ. Ferroptosis);
- Піроптоз (англ. Pyroptosis);
- Партанатоз (англ. Parthanatosis);
- Ентоз (англ. Entosis);
- NETоз (англ. NETosis).
Залежна від лізосом клітинна загибель
Залежна від лізосом клітинна загибель починається з порушень клітинного гомеостазу і пермеабілізаціі (змінення проникності) мембран лізосом. Вона спостерігається при багатьох патофізіологічних процесах: запаленні, перебудові тканин (наприклад, перебудові тканини молочних залоз після лактації), старінні, нейродегенеративних захворюваннях, серцево-судинних захворюваннях і відповіді на внутрішньоклітинні патогени[4].
Після пермеабілізаціі мембран лізосом вміст останніх виходить в цитозоль, де зокрема виявляються в результаті протеолітичні ферменти родини катепсинів, які руйнують вміст клітини. Процеси, що передують пермеабілізаціі лізосомальних мембран і запускають її, не цілком ясні. В деяких умовах вона відбувається після пермеабілізації зовнішньої мітохондріальної мембрани в ході внутрішнього апоптозу. В інших випадках пермеабілізація мембран лізосом відбувається раніше від мембран мітохондрій за участю білка BAX. Важливу роль у запуску підвищення проникності лізосомальних мембран відіграють активні форми кисню[4].
Клітинна загибель, залежна від автофагії
Залежна від автофагії клітинна загибель передбачає активацію молекулярних механізмів автофагії (всіх або частини), які призводять до утворення автофагосом — везикул з подвійною мембраною[5][6]. Автофагія є важливим процесом, що становлять частину клітинної відповіді на стрес, тому її порушення призводять до різноманітних дефектів розвитку і захворювань. У дрозофіли автофагія задіяна в оновленні вистилки середньої кишки і деградації личинкових слинних залоз. Залежна від автофагії клітинна загибель вносить свій внесок у патогенез ряду захворювань і у людини. Наприклад, при деяких патологічних станах шляхом автофагії гинуть нейрони. Різновид залежною від автофагії клітинної загибелі, в якій задіяна Na+/K+-АТФаза, відома як автоз[4].
Імуногенна клітинна загибель
Імуногенною клітинною загибеллю називають ті види клітинної смерті, які супроводжуються активацією адаптивної імунної відповіді, спрямованого проти ендогенних (клітинних) або екзогенних (вірусних) антигенів, які експресує, клітина, що гине. Імуногенну клітинну загибель викликають порівняно небагато факторів: вірусні інфекції, деякі протиракові препарати (наприклад, антрацикліни і бортезоміб]), деякі види радіотерапії, а також фотодинамічна терапія, заснована на гіперіцині. Як правило, імунну відповідь запускають такі асоційовані з ушкодженнями молекулярні патерни (англ. Damage-associated molecular pattern, DAMP), які експресуються клітиною, що вмирає: кальретикулін, АТФ, білок HMGB1, інфтерферони I типу, нуклеїнові кислоти, що походять від ракових клітин, і аннексин A1[4].
Внутрішній апоптоз
.svg.png.webp)
Внутрішній апоптоз запускають різноманітні зміни навколишнього середовища клітини: відсутність факторів росту, пошкодження ДНК, стрес ендоплазматичного ретикулуму (ЕПР), активні форми кисню, порушення реплікації ДНК, дефекти мітозу і порушення функціювання мікротрубочок. Клітини, що гинуть шляхом апоптозу, зберігають цілісність плазматичної мембрани і певну метаболічну активність. Вони розпадаються на везикули — апоптотичні тільця, які фагоцитуються іншими клітинами. Критичний етап внутрішнього апоптозу — необоротна пермеабілізація зовнішніх мітохондріальних мембран, яка контролюється різними білками родини BCL2. В результаті у цитозоль виходять проапоптотичні фактори, які в звичайний час знаходяться в міжмембранному просторі мітохондрій. Найважливішим з них є білок дихального ланцюга цитохром c. В цитозолі він зв'язується з білком APAF1 і про-каспазою 9, формуючи комплекс, відомий як апоптосома. В ній каспаза 9 активується, формуючи димери, які самі себе розрізають і тим самим активують, і починає активувати інші каспази, вносячи в них розрізи. Каспази — це протеази, які руйнують всі білки клітини і викликають смерть клітини[4].
Зовнішній апоптоз
Зовнішній апоптоз запускається змінами в мікрооточенні клітини. Ключову роль у запуску зовнішнього апоптозу відіграють два типи рецепторів клітинної мембрани: рецептори смерті, які активуються при зв'язуванні з лігандом, а також рецептори, які активуються, коли концентрація їх ліганду опускається нижче від певного значення. До числа рецепторів смерті відносяться, наприклад, рецептор Fas та низка інших рецепторів надродини факторів некрозу пухлин (англ. tumor necrosis factor, TNF). Коли рецептор смерті активується, у його внутрішньоклітинній частині збирається особливий багатобілковий комплекс — DISC (від англ. death-inducing silencing complex). Він регулює активацію і функціювання каспази-8 (або, в деяких випадках, каспази-10). Слідом за нею активуються й інші каспази, які руйнують клітинні білки і призводять до її загибелі[4].
Некроз, залежний від проникності мітохондрій
MPT-залежний некроз починається при особливих змінах внутрішньоклітинних умов, таких як сильний окислювальний стрес і значне підвищення концентрації іонів кальцію в цитозолі. Скорочення MPT походить від англ. mitochondrial permeability transition — порушення проникності мітохондрій, оскільки при цьому виді клітинної загибелі внутрішня мітохондріальна мембрана стає проникною для малих молекул, що призводить до зникнення електрохімічного градієнта на ній, осмотичного руйнування обох мітохондріальних мембран і врешті загибелі клітини у вигляді некрозу[4].
Некроптоз
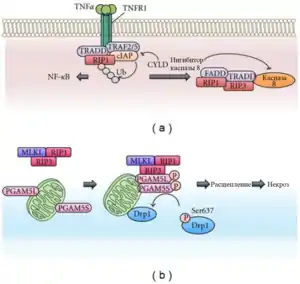
Некроптоз викликається різними змінами у внутрішньому і зовнішньому середовищі клітини, які детектуються особливими рецепторами смерті (наприклад, Fas), рецепторами розпізнавання патогенів (наприклад, Toll-подібними рецепторами 3 і 4), а також білком ZBP1, що зв'язується з Z-ДНК. Морфологічно смерть клітини відбувається у формі некрозу. Критично важливу роль у запуску некроптозу грають протеїнкінази RIPK3 і MLKL, які активуються рецепторами. Некроптоз не тільки пов'язаний з відповіддю організму на стрес, він забезпечує загибель дефектних організмів до народження і бере участь у регуляції гомеостазу T-лімфоцитів у дорослому організмі[4].
Фероптоз
Фероптозу, як правило, передує серйозне пошкодження клітинних ліпідів внаслідок утворення активних форм кисню і появи вільних йонів заліза в клітині. Окиснення ліпідів відбувається саме через йони заліза, через що даний вид клітинної загибелі і отримав свою назву[7]. Морфологічно ферроптоз — це некроз, при якому відбуваються серйозні пошкодження мітохондрій: вони стискаються, у них зникають крісти і руйнується зовнішня мембрана. У ферроптозі не задіяні каспази і білки, що здійснюють автофагію. При цьому виді клітинної загибелі відбувається окислення деяких поліненасичених жирних кислот, наприклад, арахідонової кислоти, утворюються гідропероксиди ліпідів. Іноді окиснення ліпідів може відбуватися під дією ферментів ліпоксигеназ і циклооксигеназ. Їм протидіє глутатіонпероксидаза GPX4. Ферроптозу також перешкоджають ферростатин-1, ліпрокстатин-1, а також вітамін E, коензим Q10 і подібні сполуки з антиоксидантною активністю, які відволікають на себе активні форми кисню і не дають їм взаємодіяти з ліпідами[4].
Піроптоз
Піроптоз активується в ході реакцій вродженого імунітету. При піроптозі відбувається особлива конденсація хроматину, що відрізняється від конденсації хроматину при апоптозі. Клітина розбухає, відбувається пермеабілізація мембрани. У піроптозі провідну роль відіграє прозапальна каспаза 1, однак у деяких випадках замість неї виступають інші каспази, наприклад, каспаза 3. Піроптоз задіяний у розвитку багатьох патологічних станів, наприклад, смертельного септичного шоку, спричиненого бактерійними ліпополісахаридами. Саме бактеріальні ліпополісахариди, що потрапляють у цитоплазму клітин організму-господаря, ймовірно, відіграють провідну роль у запуску піроптозу[4].
Партанатоз
Партанатоз — це форма програмованої клітинної загибелі, що характеризується гіперактивацією полі(АДФ-рибоза)-полімерази PARP1 — білка, що бере участь у клітинній відповіді на пошкодження ДНК. Однак партанатоз може відбуватися не тільки при сильному пошкодженні ДНК шляхом алкілювання, а також при окисному стресі, гіпоксії, гіпоглікемії або запаленні. Головну роль у гіперактивації PARP1, особливо в нейронах, відіграють активні форми азоту, зокрема оксид азоту NO. Гіперактивація PARP1 має цитотоксичні ефекти, такі як виснаження пулу NAD+ і АТФ (що призводить до порушення біоенергетичної і окислювально-відновної рівноваги клітини), а також накопичення полімерів полі(АДФ-рибози) і полі(АДФ-рибоз)ованих білків у мітохондріях (через що втрачається мембранний потенціал і пермеабілізується зовнішня мітохондріальна мембрана)[4].
Ентоз
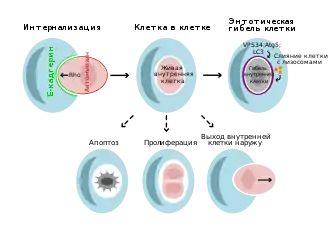
Ентоз — це форма клітинного канібалізму, яка відбувається в здорових тканинах та пухлинах ссавців. Жива клітина поглинається іншою клітиною, що не володіє здатністю до фагоцитозу. Часто, але не завжди, поглинена клітина гине. Як правило, ентоз запускається, коли епітеліальна клітина втрачає контакт з позаклітинним матриксом, хоча для цього можуть бути й інші причини: невідрегульована експресія міозинів при формуванні міжклітинних контактів, відмінності в механічних властивостях сусідніх клітин і метаболічний стрес. У ракових клітин ентоз може запускатися при мітозі. Загибель поглиненої клітини не залежить від каспаз і білків BCL2, що відіграють найважливішу роль в апоптозі. Принаймні в деяких випадках загибель відбувається у вигляді особливої форми автофагії[4].
NETоз
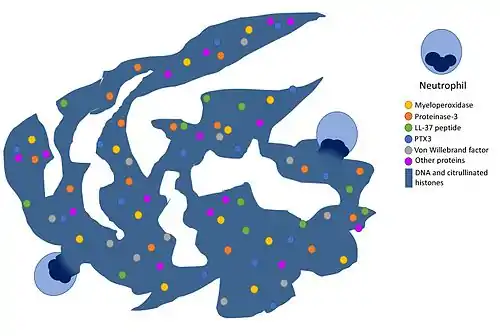
Спочатку ця форма загибелі була описана у нейтрофілів, які, вмираючи, викидають назовні мережу з волокон, що містять хроматин і гістони, пов'язані з цитозольними білками. Ці волокна були названі англ. neutrophil extracellulae traps (NET), і форма смерті отримала назву NETоз (англ. NETosis). Викид NET може бути викликаний мікробами, активацією особливих рецепторів (наприклад, Toll-подібних). Суттєва частка ДНК, яка входить до складу цих волокон, має мітохондріальне, а не ядерне походження. NET можуть викидати й інші клітини, відмінні від нейтрофілів: мастоцити, еозинофіли і базофіли, причому викид NET не завжди призводить до загибелі клітини. NET володіють не тільки антимікробним ефектом; показана їх роль у таких захворювань, як діабет і рак[4].
У безхребетних
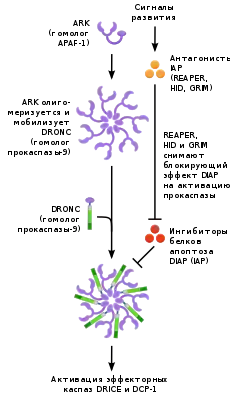
Програмована клітинна загибель зафіксована у безхребетних тварин, зокрема, у губок. У губок експресуються такі білки ПКЗ, як каспази, білки з доменом смерті і Bcl-2. Більше того, Bcl-2 губок пригнічує апоптоз і в клітинах хребетних[8]. Спочатку молекулярні механізми ПКЗ були описані у нематоди Caenorhabditis elegans. Оскільки кількість клітин у тілі дорослого хробака жорстко фіксована і однакова для всіх особин, кількість актів ПКЗ також фіксована: в ході розвитку хробака клітини вбивають самі себе рівно 131 раз. Ключову роль у ПКЗ у C. elegans грають білки Ced-4 і Ced-3 з каспазною активністю. У звичайних умовах Ced-4 пригнічений білком Ced-9, локалізованим у зовнішній мітохондріальній мембрані. Коли клітина отримує ззовні сигнал до початку ПКЗ, Ced-9 інактивується, а Ced-4 активується і в свою чергу активує Ced-3, який запускає роботу протеаз і нуклеаз[9]. У членистоногих ПКЗ вперше відбувається при утворенні нервової системи, коли відбувається диференціювання і ділення клітин ектодерми, причому одна з дочірніх клітин стає нейробластом, а інша гине[10]. Більше того, внаслідок ПКЗ у самців і самок деякі органи іннервуються по-різному[11]. У плодової мушки Drosophila melanogaster є кілька каспаз та інгібіторів апоптозу, крім того, деякі білки ПКЗ, такі як REAPER, HID і GRIM, можуть бути специфічні для комах[12].
У рослин
У рослин програмована клітинна загибель спостерігається при утворенні ксилеми і насіння, старінні, запобіганні самозапилення, а також під дією стресів (сольового, температурного, окисного) і патогенів. Як і у тварин, у рослин існує кілька видів ПКЗ, проте найчастіше вона схожа з апоптозом і супроводжується фрагментацією ДНК, виходом цитохрому c з мітохондрій, стисненням клітини, утворенням активних форм кисню і виходом фосфатидилсерину на зовнішній шар мембрани. Разом з тим, хоча у рослин відсутні каспази, відомо, що інгібітори каспаз тварин можуть пригнічувати програмовану клітинну смерть і у рослин. Головна роль програмованої клітинної загибелі у рослин належить фітаспазам — серин-залежним аспартат-специфічним протеазам. У здорових тканинах фітаспази знаходяться в апопласті, а при індукції ПКЗ входять у цитозоль[1].
У грибів
У грибів програмована клітинна загибель спостерігається при утворенні спор статевого і безстатевого розмноження, формуванні плодового тіла або склероцію, в реакції вегетативної несумісності, при патогенезі, стресових умовах і на заключних етапах старіння. Цим призначення ПКЗ у грибів відрізняється від такої у тварин, у яких вона перш за все важлива для розвитку. У загальному випадку ПКЗ грибів аналогічна внутрішньому апоптозу тварин. ПКЗ детально вивчена у дріжджів Saccharomyces cerevisiae і може запускатися різноманітними внутрішніми факторами, причому зовнішній механізм активації ПКЗ не виявлено. У них немає і очевидних гомологів ключових білків апоптозу тварин, таких як Bcl-2, p53, FLIP, PARP і навіть каспази. У той же час гомологи деяких регуляторних апоптотичних білків відсутні у дріжджів, але є у міцеліальних грибів. У Podospora anserina ПКЗ проявляється при старінні міцелію, яке зумовлене дією активних форм кисню. В ході ПКЗ у P. anserina функціонують цистеїнові протеази з каспазною активністю[2].
У слизовиків
Плодове тіло слизовика Dictyostelium discoideum має ніжку, утворену мертвими клітинами. Ці клітини зазнали ПКЗ, схожої на автофагію тварин за ступенем розвитку вакуоль і конденсації хроматину, крім того, на відміну апоптозу, фрагментації ДНК не відбувається[13]. Предки слизовиків відокремилися від інших еукаріотів більше мільярда років тому до відділення предків рослин і грибів, що свідчить про давнє походження програмованої клітинної загибелі[14].
У бактерій
У бактерій відомо кілька форм програмованої клітинної загибелі. В умовах стресу (окисного стресу, впливу радіації, нестачі поживних речовин, фагової інфекції) частина клітин гине на благо колонії. Найчастіше смерть відбувається за участю систем токсин-антитоксин різних типів. Бактеріофаги, геном яких представлений двохланцюжковою ДНК, спричиняють загибель заражених клітин в кінці літичного циклу для вивільнення нових віріонів з допомогою голін-ендолізинової системи. Маленькі білки голіни вбудовуються у мембрану, даючи можливість вийти назовні ендолізинам. Ендолізини гідролізують пептидоглікан, руйнують клітинну стінку і викликають лізис клітини. Загибель бактеріальних клітин спостерігається на різних етапах розвитку колонії і за відсутності стресу: при споруляції, генетичній трансформації, утворенні плодових тіл та формуванні біоплівок. Механізми програмованої клітинної загибелі у всіх випадках різні[3].
Фізіологічне значення
Фізіологічне значення програмованої клітинної загибелі величезне. У тварин вона відіграє найважливішу роль у розвитку багатьох органів і тканин, а також старінні. У ході розвитку нервової системи безліч клітин-попередників нейронів гинуть, так що кількість нейронів у мозку дорослої тварини істотно менша, ніж їх було закладено в ході ембріонального розвитку. Апоптоз задіяний у морфогенезі тварин (зокрема, апоптозом гинуть клітини між пальцями, за рахунок апоптозу відпадає хвіст у пуголовка). Імуногенна клітинна смерть і піроптоз поряд з апоптозом задіяні в роботі захисних систем організму. Пригнічення ПКЗ дуже часто пов'язане із злоякісним переродженням клітини[15]. У рослин ПКЗ бере участь в утворенні тканин, що складаються з мертвих клітин, наприклад, ксилеми. Крім того, на ПКЗ заснована самонесумісність при запиленні: якщо на рильце потрапляє пилок від тієї ж рослини, то особливі білки на рильці запускають ПКЗ клітин пилкового зерна[16]. У грибів ПКЗ забезпечує вегетативну несумісність, тобто не дає зливатися гіфам одного штаму, а також задіяна у дозріванні спор статевого і безстатевого розмноження[2].
Історія вивчення
.jpg.webp)
Сама концепція програмованої клітинної загибелі була запропонована Локшином (англ. Lockshin) і Вільямсом у 1964 році стосовно розвитку деяких тканин у комах[17]. Приблизно через 8 років з'явився термін «апоптоз». Перші відомості про механізми ПКЗ з'явилися при вивченні білка Bcl-2 — продукту онкогену, експресія якого активується при хромосомних транслокаціях, які часто спостерігаються при фолікулярній лімфомі. На відміну від інших відомих до цього моменту продуктів онкогенів, Bcl-2 викликає злоякісне переродження не за рахунок безперервної стимуляції поділу, а за рахунок запобігання програмованої клітинної загибелі[18]. Донині програмована клітинна загибель інтенсивно досліджується. У 2002 році Нобелівська премія з фізіології і медицини була присуджена за відкриття в молекулярній біології програмованої клітинної загибелі Сідні Бреннеру, Роберту Горвіцу і Джону Салстону[19], а в 2016 році цієї нагороди був удостоєний Йосінорі Осумі, який досліджував один із видів програмованої клітинної загибелі — автофагію[20].
Примітки
- Фомичева А. С. Программируемая клеточная смерть у растений / Тужиков А. И., Белошистов Р. Е., Трусова С. В., Галиуллина Р. А., Мочалова Л. В., Чичкова Н. В., Вартапетян А. Б. // Успехи биологической химии. — 2012. — Т. 52. — С. 97—126.
- Камзолкина О. В., Дунаевский Я. Е. Биология грибной клетки. — М. : Товарищество научных изданий КМК, 2015. — С. 217—223. — 239 с. — ISBN 978-5-9906564-1-3.
- PMID 25611384 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - PMID 29362479 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - PMID 21801009 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - PMID 17909521 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - PMID 24346035 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - DOI:10.1007/s00391-004-0230-5
Нема шаблону {{Cite doi/10.1007/s00391-004-0230-5}}.заповнити вручну - Jane B. Reece, Lisa A. Urry, Michael L. Cain, Steven A. Wasserman, Peter V. Minorsky, Robert B. Jackson. Campbell Biology. — Pearson, 2014. — С. 228. — ISBN 978-0-321-77565-8.
- PMID 4029506 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - DOI:10.1002/cne.902260107
Нема шаблону {{Cite doi/10.1002/cne.902260107}}.заповнити вручну - PMID 10908589 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - PMID 12654899 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - PMID 14681218 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - Ярилин А. А. Апоптоз и его роль в целостном организме // Глаукома. — 2003. — Вип. 2. — С. 46—54.
- PMID 15152254 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - DOI:10.1016/0022-1910(64)90034-4
Нема шаблону {{Cite doi/10.1016/0022-1910(64)90034-4}}.заповнити вручну - PMID 3262202 (PubMed)
Бібліографічний опис з'явиться автоматично через деякий час. Ви можете підставити цитату власноруч або використовуючи бота. - The Nobel Prize in Physiology or Medicine 2002. The Nobel Foundation. 2002. Процитовано 21 червня 2009.
- The Nobel Prize in Physiology or Medicine 2016. The Nobel Foundation. 3 жовтня 2016. Процитовано 3 жовтня 2016.