Т-хелпер
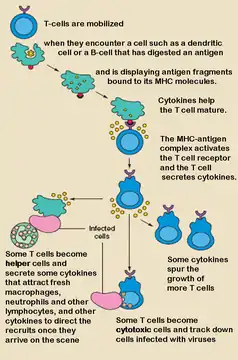
Т-клітини-хелпери (Th-клітини), також відомі як, Т-хелпери, CD4+-клітини або CD4-позитивні клітини — тип Т-клітин, які відіграють важливу роль в імунній системі, зокрема в адаптивній імунній системі. Як випливає з назви, вони «допомагають» активності інших імунних клітин, вивільняючи цитокіни, невеликі білкові медіатори, які змінюють поведінку клітин-мішеней, які експресують рецептори цих цитокінів. Ці клітини допомагають поляризувати (спрямувати) імунну відповідь у відповідний вид залежно від природи імунологічного ураження (вірус, чи позаклітинна бактерія, чи внутрішньоклітинна бактерія, чи гельмінт, чи грибок, чи протиста). Зазвичай вони вважаються важливими для перемикання класу антитіл В-клітин, порушення перехресної толерантності дендритних клітин, для активації та росту цитотоксичних Т-клітин, а також для максимального підвищення бактерицидної активності фагоцитів, таких як макрофаги та нейтрофіли.
| Т-хелпер | |
| Код за Foundational Model of Anatomy | 70572[1] |
|---|---|
 | |
| | |
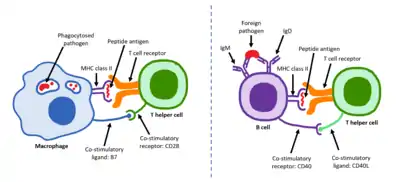
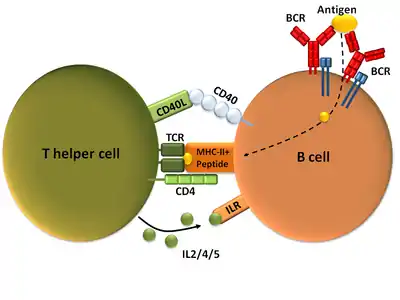
Зрілі Th-клітини експресують поверхневий білок CD4 і відомі як CD4+ Т-клітини. Такі CD4+ Т-клітини зазвичай розглядаються як такі, що мають заздалегідь визначену роль клітин-помічників в імунній системі. Наприклад, коли антигенпрезентувальна клітина демонструє пептидний антиген на білках MHC класу II, клітина CD4+ допомагає цим клітинам через комбінацію взаємодій між клітинами (наприклад, CD40 (білок) і CD40L) і через цитокіни.
CD154, також званий лігандом CD40 або CD40L, є білком клітинної поверхні, який опосередковує функцію Т-хелперів у контактно-залежному процесі[2] і є членом суперсімейства молекул TNF. Він зв'язується з CD40 на антигенпрезентувальних клітинах (АПК), що призводить до багатьох ефектів залежно від типу клітини-мішені. CD154 діє як костимулююча молекула і особливо важливий для підгрупи Т-клітин, яка називаються фолікулярні Т-хелпери (ТFH).[3] На TFH клітинах CD154 сприяє дозріванню та функціонуванню B-клітин, залучаючи CD40 на їх поверхні і, отже, полегшуючи комунікацію між клітинами.[4] Дефект цього гена призводить до нездатності проводити перемикання класу імуноглобулінів і асоціюється з синдромом гіпер-IgM.[5] Відсутність CD154 також зупиняє утворення зародкових центрів і, отже, забороняє дозрівання спорідненості антитіл, важливого процесу в адаптивній імунній системі, який дозволяє генерувати високоафінні антитіла, які захищають від інфікування у майбутньому.
У сукупності важливість Т-хелперів можна побачити при інфекції ВІЛ, вірусу, який в першу чергу інфікує CD4+ Т-клітини (але здатний інфікувати інші важливі клітини імунної системи, наприклад макрофаги, які теж експресують CD4). На запущених стадіях ВІЛ-інфекції втрата функціональних CD4+ Т-клітин призводить до симптомної стадії інфекції, відомої як синдром набутого імунодефіциту (СНІД). Коли ВІЛ виявлено на ранніх стадіях в крові або інших рідинах організму, належне дотримання антиретровірусної терапії запобігає переростання ВІЛ у СНІД і дозволяє організму природним чином відновити кількість власних CD4-клітин (невелика частина людей здатна досягти тривалого контролю вірусного навантаження без значного зниження рівня Т-клітин з часом без допомоги антиретровірусної терапії. Їх називаються «елітними контролерами» або «довготривалими непрогресорами»). ВІЛ є прикладом вторинного імунодефіциту. Первинні імунодефіцити — це генетичні стани, які призводять до імунологічних дефектів, що перешкоджають адекватній боротьбі з інфекціями, а дефіцит Т-клітин є особливо руйнівним. Одним із прикладів є SCID (важкий комбінований імунодефіцит), який має багато причин і різниться у своєму точному фенотипі залежно від причини. Хоча більшість форм SCID призводять до відсутності як Т-, так і В-клітинної лінії, атиповий SCID характеризується (частково) нормальним рівнем В-клітин, але глибоким дефіцитом Т-клітин. Однак, через дефіцит Т-хелперів, В-клітини мають глибоке порушення функції.[6]
Важливо розуміти, що розглядати Th-клітини як монолітну імунологічну сутність є помилковим, оскільки вони надзвичайно різноманітні з точки зору функцій та їх взаємодії з клітинами-партнерами (про це детальніше йдеться нижче). Загалом, зрілі наївні Т-клітини (ті, які пройшли через контрольні точки розвитку в тимусі, але ще не зустріли свій споріднений антиген) мають бути активовані професійними антигенпрезентувальними клітинами, щоб отримати ефекторний модуль. Вони визначаються наявністю фактора транскрипції, що визначає їх родовід (або специфікує лінію), (також званого головним регулятором, хоча цей термін критикували як занадто спрощений).[7] Втрата функції в лінії, що визначає транскрипційний фактор, призводить до відсутності відповідного класу Т-хелперів, що може бути руйнівним для здоров'я хазяїна. Наприклад, звичайні регуляторні Т-клітини (Treg-клітини) визначаються експресією FoxP3 (Forkhead Box P3), і втрата функції цього транскрипційного фактора призводить до стану, який називається IPEX (імунодисрегуляційна поліендокринопатія, ентеропатія, Х-зчеплена), синдром який характеризується неконтрольованими імунними реакціями, які спричиняють серйозне пошкодження тканин, а також блискавичним аутоімунним ураженням (зауважте, що це аутоімунне захворювання, а не лише наявність аутореактивності), оскільки потрібна популяція Т-хелперів відсутня. У деяких випадках мутація, що веде до втрати функції, може відбутися вище від транскрипційного фактора, що специфікує лінію. Наприклад, при синдромі гіпер-IgE є мутація в гені STAT3, який відповідає за індукцію фактора транскрипції RORγT у відповідь на комбінацію TGF-β та або IL-6, або IL-21, що визначає клітини TH17. Внаслідок відсутності цих клітин пацієнти мають серйозні грибкові інфекції та важко реагують на гнійні бактеріальні патогени (хоча вони також мають інші імунологічні дефекти, оскільки STAT3 також бере участь в інших сигнальних шляхах).[8] Дефіцит STAT3 також ставить під загрозу здатність генерувати ТFH-клітини.[9]
Активація наївних Т-хелперів
Після розвитку Т-клітин у вилочковій залозі ці клітини (так звані «нещодавні емігранти з тимуса» — RTE) виходять із тимуса і заселяють вторинні лімфоїдні органи (ВЛО; селезінка та лімфатичні вузли). Слід зазначити, що лише дуже невелика частина Т-клітин виходить із тимуса (за оцінками зазвичай коливається від 1-5 %, але деякі експерти вважають навіть таку оцінку дуже щедрою).[10] Дозрівання RTE у ВЛО призводить до появи наївних Т-клітин (наївні означає, що вони ніколи не були в контакті з антигеном, з яким вони запрограмовані реагувати), але у наївної Т-клітини тепер відсутні або зменшена експресія поверхневих молекул-маркерів, характерних для RTE, такі як CD31, PTK7, рецептори комплементу 1 і 2 (CR1, CR2) та продукування інтерлейкіну 8 (IL-8).[11][12]
Як і всі Т-клітини, вони експресують Т-клітинний рецептор — комплекс CD3. Т-клітинний рецептор (TCR) складається з константних і варіабельних ділянок. Варіабельна ділянка визначає, на який антиген може реагувати Т-клітина. CD4+ Т-клітини мають TCR зі спорідненістю до MHC класу II, а CD4 бере участь у визначенні спорідненості MHC під час дозрівання в тимусі. Білки MHCII зазвичай зустрічаються лише на поверхні професійних антигенпрезентувальних клітин (АПК). Професійні АПК — це переважно дендритні клітини, макрофаги та В-клітини, хоча дендритні клітини є єдиною групою клітин, яка конститутивно (завжди) експресує MHCII. Деякі АПК також зв'язують нативні (або необроблені) антигени зі своєю поверхнею, наприклад, фолікулярні дендритні клітини (це не той самий тип клітин, що й дендритні клітини імунної системи, і вони мають негематопоетичне походження і загалом не мають MHCII, тобто вони не є справжніми професійними АПК; однак фолікулярні дендритні клітини можуть отримувати білки MHCII через екзосоми, які приєднуються до них[13]).
Т-клітини потребують процесінгу антигенів у короткі фрагменти, які утворюють лінійні епітопи на МНСII (у випадку Т-хелперів, оскільки вони експресують CD4) або МНСI (у випадку цитотоксичних Т-клітин, які експресують CD8). Зв'язувальні кишені MHCII є гнучкими щодо довжини пептидів, які вони містять. Як правило, існує 9 основних амінокислотних залишків з кількома додатковими амінокислотами, які утворюють довжину приблизно 12-16 амінокислот[14] але, також відомі ті, що містять до 25 амінокислот.[15] Для порівняння, білки MHC класу I зазвичай мають довжину 9-10 пептидів.[16] Активація наївних Т-клітин зазвичай пояснюється з точки зору 3-сигнальної моделі, детально розглянутої нижче.[17]
Активація (сигнал 1)
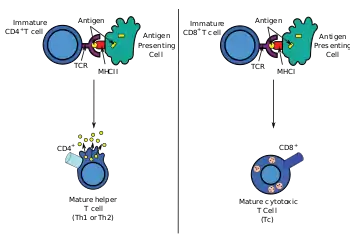
Під час імунної відповіді професійні АПК поглинають антигени (як правило, бактерії або віруси), які піддаються обробці, а потім АПК переміщуються від місця інфекції до лімфатичних вузлів. Як правило, відповідальною АПК є дендритна клітина. Якщо антиген експресує відповідні молекулярні структури (іноді відомі як сигнал 0), він може викликати дозрівання дендритної клітини, що призводить до посилення експресії костимулюючих молекул, необхідних для активації Т-клітин (див. сигнал 2)[18] і MHC класу II.[19] Потрапляючи в лімфатичні вузли, АПК починають представляти антигенні пептиди, які зв'язані з MHC класу II, що дозволяє активувати CD4+ T-клітини, які експресують специфічні TCR проти комплексу пептид/MHC.
Коли Th- клітина зустрічає і розпізнає антиген на АПК, комплекс TCR-CD3 міцно зв'язується з комплексом пептид-MHC, присутнім на поверхні професійних АПК. CD4, корецептор комплексу TCR, також зв'язується з іншою частиною молекули MHC. Підраховано, що для активації Т-хелпера неохідно приблизно 50 цих взаємодій. В зоні контакту спостерігалося формування сукупностей (мікрокластери) між комплексами TCR-CD3-CD4 Т-клітини та білками MHC класу II дендритної клітини. Коли все це об'єднується, CD4 здатний залучити кіназу під назвою Lck, яка фосфорилює мотиви активації імунотирозину (ITAM), присутні на гамма-, дельта-, епсилон- та дзета-ланцюгах CD3. Білок ZAP-70 може зв'язувати ці фосфорильовані ITAM через свій домен SH2,а потім сам стає фосфорильованим, при цьому він організовує передачу сигналів, необхідну для активації Т-клітин. Активація Lck контролюється протилежними діями CD45 і Csk.[20] CD45 активує Lck шляхом дефосфорилювання тирозину в його С-кінцевому хвості, тоді як Csk фосфорилює Lck на цьому місці. Втрата CD45 утворює форму SCID, оскільки нездатність активувати Lck перешкоджає передачі сигналу Т-клітинами. Т-клітини пам'яті також використовують цей шлях і мають більш високий рівень експресії Lck, а функція Csk у цих клітинах пригнічується.[21]
Зв'язування комплексу антиген-МНС з TCR і CD4 може також допомогти з'єднанню АПК і Th при активації останніх. Білок інтегрин LFA-1, на Т-клітині та ICAM на АПК є основними молекулами адгезії в цій взаємодії.
Невідомо, яку роль відіграє масивна ділянка CD45 під час клітинних взаємодій, але ця молекула має різні ізоформи, що змінює розмір в залежності від стану активації і дозрівання Th. Наприклад, CD45 скорочується в довжині після активації Th (з CD45RA+ до CD45RO+), але чи впливає ця зміна довжини на активацію, невідомо. Було припущено, що більший CD45RA може зменшити доступність Т-клітинного рецептора для комплексу антиген-MHC, тим самим вимагаючи збільшення спорідненості (і специфічності) Т-клітини для активації Th. Однак після активації CD45 скорочується, що дозволяє легше взаємодіяти та активувати Th.
Виживання (сигнал 2)
Отримавши перший сигнал TCR/CD3, наївна Т-клітина повинна активувати другий незалежний біохімічний шлях, відомий як Сигнал 2. Цей етап верифікації є захисним заходом, щоб переконатися, що Т-клітина реагує на чужорідний антиген. Якщо цей другий сигнал відсутній під час початкового впливу антигену, Т-клітина «припускає», що вона автореактивна. Це призводить до того, що клітина стає анергичною (анергія утворюється внаслідок незавершених біохімічних змін після Сигналу 1). Анергічні клітини не будуть реагувати на жодний антиген у майбутньому, навіть якщо обидва сигнали будуть присутні пізніше. Вважається, що ці клітини циркулюють по всьому тілу без жодної цінності, поки вони не піддадуться апоптозу.
Другий сигнал включає взаємодію між CD28 на CD4+ Т-клітині та білками CD80 (B7.1) або CD86 (B7.2) на професійних АПК. І CD80, і CD86 активують рецептор CD28. Ці білки також відомі як костимуляторні молекули.
Хоча етап верифікації необхідний для активації наївних хелперних Т-клітин, його важливість було найкраще продемонстровано під час подібного механізму активації CD8+ цитотоксичних Т-клітин. Оскільки наївні CD8+ Т-клітини не мають справжньої схильності щодо чужорідних речовин, вони повинні покладатися на активацію CD28 для підтвердження того, що вони розпізнають чужорідний антиген (оскільки CD80/CD86 експресується лише активними АПК). CD28 відіграє важливу роль у зниженні ризику аутоімунітету Т-клітин проти антигенів хазяїна.
Після того, як у наївної Т-клітини активуються обидва шляхи, біохімічні зміни, викликані Сигналом 1, змінюються, що дозволяє клітині активуватися замість того, щоб ставати анергічною. Це також вірно для Т-клітин пам'яті, які є одним із прикладів адаптивного імунітету. При повторному зараженні реакції відбуваються швидше, оскільки Т-клітини пам'яті вже пройшли верифікацію і можуть виробляти ефекторні клітини набагато раніше.
Диференціація (сигнал 3)
Після того, як активація двома сигналами завершена, Т-хелпер може розмножуватися. Це досягається шляхом вивільнення потужного фактора росту Т-клітин, який називається інтерлейкін 2 (IL-2), який сам діє на свою клітину аутокринним чином. Активовані Т-клітини також виробляють альфа-субодиницю рецептора IL-2 (CD25 або IL-2R), створюючи повністю функціональний рецептор, який може зв'язуватися з IL-2, що, у свою чергу, активує шляхи проліферації Т-клітин.
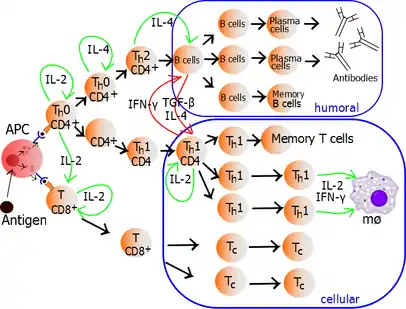
Аутокринно або паракринно вивільнений IL-2 може зв'язуватися з цією ж Th-клітиною або сусідніми Th через IL-2R, таким чином стимулюючи проліферацію та клональну експансію. Th, який отримує як сигнали активації і проліферації стане тоді Th0 (Т-хелпер 0), який секретує IL-2, IL-4 і гамма-інтерферон (IFN-γ). Потім Th0 диференціюється на клітини Th1 або Th2 залежно від середовища цитокінів. IFN-γ стимулює продукцію Th1, тоді як IL-10 та IL-4 пригнічують продукцію Th1. З іншого боку, ІЛ-4 призводить до продукування Th2, а IFN-γ пригнічує появу Th2. Ці цитокіни є плейотропними і виконують багато інших функцій імунної відповіді.
Ефекторна функція
У 1991 році три групи дослідників повідомили про відкриття CD154, який є молекулярною основою хелперної функції Т-клітин. Сет Ледерман з Колумбійського університету виділив мишаче моноклональне антитіло 5c8, яке інгібує контактно-залежну функцію Т-хелперів у клітинах людини, що характеризує поверхневий білок з масою 32 кДа, який тимчасово експресується на CD4+ Т-клітинах.[2] Річард Армітаж з компанії Immunex клонував кДНК, що кодує CD154, скринінгуючи бібліотеку експресії за допомогою CD40-Ig.[22] Рендольф Ноель з Дартмутської медичної школи створив антитіло, яке зв'язувало білок молекулярної маси 39 кДа на мишачих Т-клітинах і пригнічував функцію хелперів.[23]
Визначення ефекторної функції Т-клітин
Т-хелпери здатні впливати на різноманітні імунні клітини, і їх реакції (включаючи позаклітинні сигнали, такі як цитокіни) можуть бути важливими для успішної боротьби з інфекцією. Щоб стати ефекторними, Т-хелпери повинні «визначити», які цитокіни дозволять імунній системі бути найбільш дієвою для організму господаря. Розуміння того, як Т-хелпери реагують на імунні виклики, в даний час представляє великий інтерес в імунології, оскільки такі знання можуть бути дуже корисними для лікування захворювань і підвищення ефективності вакцинації.
Модель Th1/Th2
Проліферуючі Т-хелпери, які розвиваються в ефекторні Т-клітини, диференціюються на два основних підтипи клітин, відомих як Тh1 і Тh2 (також відомі як Т-хелпери типу 1 і типу 2 відповідно).
Тh1 призводить до посилення клітинного імунітету (в першу чергу, реакцій макрофагів і цитотоксичних Т-клітин)[24], як правило, проти внутрішньоклітинних бактерій і простіших. Ці реакції запускаються поляризаційним цитокіном IL-12, а їх ефекторними цитокінами є IFN-γ та IL-2. Основними ефекторними клітинами імунітету Тh1 є макрофаги, а також цитотоксичні Т-клітини, IgG B-клітини та IFN-γ CD4+ Т-клітини. Ключовими факторами транскрипції Тh1 є STAT4 і T-bet. IFN-γ, що секретується Т-хелперами, може активувати макрофаги для фагоцитозу та перетравлення внутрішньоклітинних бактерій і простіших. Крім того, IFN-γ може активувати iNOS (синтазу оксиду азоту, що індукується) для продукції вільних радикалів оксиду азоту для безпосереднього знищення внутрішньоклітинних бактерій і простіших. Гіперактивація Тh1 проти аутоантигенів спричиняє реакцію гіперчутливості IV типу або реакцію уповільненого типу. До цієї категорії реакцій гіперчутливості належать туберкулінова реакція або цукровий діабет 1 типу.[25]
Тh2 призводять до гуморальної імунної відповіді[24], як правило, проти позаклітинних паразитів, таких як гельмінти. Вони запускаються поляризаційними цитокінами IL-4 та IL-2, а їх ефекторними цитокінами є IL-4, IL-5, IL-9, IL-10, IL-13 та IL-25. Основними ефекторними клітинами є еозинофіли, базофіли та тучні клітини, а також В-клітини та IL-4/IL-5 CD4 Т-клітини. Ключовими факторами транскрипції Тh2 є STAT6 і GATA3.[26] IL-4 є цитокіном позитивного зворотного зв'язку для диференціювання клітин Тh2. Крім того, IL-4 стимулює В-клітини виробляти антитіла IgE, які, в свою чергу, стимулюють тучні клітини вивільняти гістамін, серотонін і лейкотрієн, що викликає звуження бронхів, посилену перистальтику кишечника, підкислення шлункового соку для вигнання гельмінтів. ІЛ-5 із CD4 Т-клітин активує еозинофіли для нападу на гельмінтів. IL-10 пригнічує диференціювання Th1 клітин і функцію дендритних клітин. Гіперактивація Th2 проти антигену спричинить гіперчутливість I типу, яка є алергічною реакцією, опосередкованою IgE. До цієї категорії гіперактивації належать алергічний риніт, атопічний дерматит та астма.[25] Окрім експресії різних цитокінів, клітини Th2 також відрізняються від клітин Th1 гліканами на клітинній поверхні (олігосахаридами), що робить їх менш сприйнятливими до деяких індукторів загибелі клітин.[27][28]
| Тип 1 (Th1) | Тип 2 (Th2)[25] | |
|---|---|---|
| Основний тип клітини-партнера | Макрофаг, CD8+ Т-клітина | В-клітини, еозинофіли, тучні клітини |
| Цитокіни, що виробляються | Інтерферон гамма (IFNγ) і TNF-β. Повідомлялося про вироблення інтерлейкіну 2 та інтерлейкіну 10 в активованих Th1.[30] | Інтерлейкін 4, інтерлейкін 5, інтерлейкін 6, інтерлейкін 9, інтерлейкін 10, інтерлейкін 13 |
| Стимуляція імунітету | Клітинний імунітет. Максимізує ефективність знищення макрофагами і проліферацію цитотоксичних Т-клітин. Також сприяє виробленню IgG, опсонізуючого антитіла. | Гуморальний імунітет. Стимулює проліферацію В-клітин, індукує зміну класу антитіл В-клітин і збільшує вироблення нейтралізуючих антитіл (IgG, IgM та IgA, а також антитіл IgE). |
| Інші функції | Цитокін Тh1 IFNγ збільшує продукцію інтерлейкіну 12 дендритними клітинами і макрофагами, а за допомогою позитивного зворотного зв'язку IL-12 стимулює продукцію IFNγ в Т-хелперах, тим самим сприяючи розвитку профілю Th1. IFNγ також пригнічує вироблення цитокінів, таких як інтерлейкін 4, важливого цитокіну, пов'язаного з реакцією Тh2, і, таким чином, зберігається власна відповідь Тh1. | Тh2 сприяє розвитку власного профілю за допомогою двох різних цитокінів. Інтерлейкін 4 сприяє виробленню цитокінів Тh2 (у тому числі самих себе; він є авторегуляторним), тоді як інтерлейкін 10 (IL-10) інгібує різноманітні цитокіни, включаючи інтерлейкін 2 та IFNγ у Тh та ІЛ-12 в дендритних клітинах і макрофагах. Комбінована дія цих двох цитокінів свідчить про те, що як тільки Т-клітина «вирішила» виробляти ці цитокіни, це рішення зберігається (і також спонукає інші Т-клітини робити те ж саме). |
Хоча ми знаємо про цитокінові патерни, характерні для Тh, ми менше розуміємо, як ці патерни визначаються. Різні дані свідчать про те, що тип АПК, який представляє антиген Т-клітині, має великий вплив на її профіль. Інші дані свідчать про те, що концентрація антигену, представленого Т-клітині під час первинної активації, впливає на те, на який підтип у подальшому вона розвинеться. Наявність деяких цитокінів (наприклад, згаданих вище) також вплине на реакцію, але наше розуміння зараз далеко не повне.
Th17 хелпери
Th17-хелпери є підгрупою T-хелперів, які відрізняються від ліній Тh1 і Th2 тим, що продукують інтерлейкін 17 (IL-17). Тh клітини виробляють ІЛ-17, який є прозапальною речовиною. Це означає, що він особливо дієвий проти позаклітинних патогенів та грибків.
THαβ хелпери
THαβ забезпечують імунітет хазяїна проти вірусів. Їх диференціювання ініціюється ІФН α/β або ІЛ-10. Їх ключовим ефекторним цитокіном є IL-10. Основними ефекторними клітинами є NK-клітини, а також цитотоксичні Т-клітини, IgG B-клітини та IL-10 CD4+ Т-клітини. Ключовими факторами транскрипції THαβ є STAT1 і STAT3, а також IRF. IL-10 із CD4 Т-клітин активує антитілозалежну клітинну цитотоксичність NK-клітин для апоптозу клітин, інфікованих вірусом, і для індукування фрагментації ДНК хазяїна та вірусу. Інтерферон альфа/бета може пригнічувати транскрипцію, щоб блокуючи реплікацію та передачі вірусу. Гіперактивація THαβ проти аутоантигену спричиняє цитотоксичну гіперчутливість типу 2, залежну від антитіл. До цієї категорії належать міастенія або хвороба Грейвса.
Обмеження моделі Th1/Th2
Взаємодія між цитокінами з моделі Th1/Th2 може бути більш складною у деяких тварин. Наприклад, цитокін Th2 IL-10 інгібує вироблення цитокінів обох підгруп Th у людей. ІЛ-10 людини (hIL-10) пригнічує проліферацію та вироблення цитокінів усіх Т-клітин і активність макрофагів, але продовжує стимулювати плазматичні клітини, забезпечуючи продукцію антитіл. Таким чином, вважається, що hIL-10 дійсно не сприяє реакції Th2 у людей, але діє, запобігаючи надмірній стимуляції Т-хелперів, одночасно максимізуючи виробництво антитіл.
Існують також інші типи Т-клітин, які можуть впливати на експресію та активацію Т-хелперів, наприклад, природні регуляторні Т-клітини, а також менш поширені профілі цитокінів, такі як підгрупа Th3. Такі терміни, як «регулятор» і «супресор», стали неоднозначними після відкриття того, що Т-хелпери також здатні регулювати (і пригнічувати) свої власні реакції за межами виокремлених регуляторних Т-клітин.
Однією з основних відмінностей між регуляторними та ефекторними Т-клітинами є те, що перші, як правило, служать для модуляції та дезактивації імунної відповіді, тоді як ефекторні Т-клітини зазвичай починають з секреції цитокінів, що стимулюють імунітет, а пізніше у своєму життєвому циклі перемикаються на цитокіни, які його гальмують. Останнє є особливістю клітин Th3, які після первинної активації та продукції цитокінів трансформуються в регуляторну підгрупу.
Як регуляторні Т-клітини, так і Th3-клітини продукують цитокін трансформуючий фактор росту-бета (TGF-β) та IL-10. Обидва цитокіни інгібують Т-хелпери; TGF-β пригнічує активність більшості компонентів імунної системи. Існують докази того, що TGF-β може не так ефективно пригнічувати активовані Th2-клітини, як він міг би пригнічувати наївні клітини, але зазвичай його не вважають цитокіном Th2.
Виокремлення іншого нового підтипу Т-хелперів — Th17[31] поставила під сумнів базову модель Th1/Th2. Ці клітини, що продукують IL-17, спочатку були описані як патогенна популяція, пов'язана з аутоімунітетом, але тепер вважають, що вони мають свої власні ефекторні та регуляторні функції. Слід зазначити, що останні дані свідчать про те, що функціональна пластичність є особливістю Т-хелперів. Справді, дослідження на мишах продемонструвало, що клітини Th17 трансформуються в клітини Th1 in vivo.[32] Подальше дослідження також показало, що велика пластичність Т-хелперів також помітна у людини.[33]
Багато цитокінів, які згадуються у цій статті, також експресуються іншими імунними клітинами, і стає зрозумілим, що хоча оригінальна модель Th1/Th2 є провідною та дає уявлення про функції T-хелпера, вона занадто проста для визначення всіх її функцій. Деякі імунологи повністю відкидають цю модель, оскільки деякі дослідження in vivo свідчать про те, що окремі Т-хелпери зазвичай не відповідають специфічним профілям цитокінів даної моделі, а багато клітин експресують цитокіни обох профілів.[34] Тим не менш, ця модель все ще відіграє важливу роль у розвитку нашого розуміння ролі та поведінки Т-хелперів і цитокінів, які вони виробляють під час імунної відповіді.
Останні наукові дослідження Stockinger et al. показали, що може існувати ще одна підмножина T-хелперів. Як вони стверджують, клітини Th9 є підгрупою Т-клітин, що продукує IL9 (інтерлейкін 9). Вважають, що ці клітини відіграють певну роль переважно при захисті від гельмінтних інфекцій.[35]
Т-клітина пам'яті
Історично вважалося, що Т-клітини пам'яті належать до ефекторного або центрального підтипу клітин пам'яті, кожен з яких має свій власний набір маркерів клітинної поверхні.[36] Центральні Т-клітини пам'яті знаходяться в лімфатичних вузлах, тоді як ефекторні Т-клітини пам'яті не мають рецепторів CC-хемокінів типу 7 (CCR7) і L-селектину (CD62L), що запобігає їх транспорту до лімфатичних вузлів.
Зараз відомо, що існують інші популяції Т-клітин пам'яті. До них належать тканинні Т-клітини пам'яті (Trm) і Т-клітини віртуальної пам'яті.[37] Єдиною об'єднуючою ознакою для всіх підтипів Т-клітин пам'яті є те, що вони довгоживучі і можуть швидко перетворюватися на велику кількість ефекторних Т-клітин при зустрічі зі спорідненим антигеном. Завдяки цьому механізму вони забезпечують імунну систему «пам'яттю» проти патогенів, які вже зустрічалися.
Роль у захворюванні
Враховуючи різноманітну та важливу роль Т-хелперів в імунній системі, не дивно, що вони часто впливають на імунну відповідь проти збудників хвороб. Вони також іноді «роблять помилки» або дають реакції, які вважаються «невигідними». У найгіршому випадку реакція Т-хелперів може призвести до катастрофи і смерті хазяїна. На щастя, це дуже рідкісне явище.
Протипухлинний імунітет
Гіперчутливість
Імунна система повинна мати баланс чутливості, щоб реагувати на чужорідні антигени, не реагуючи на антигени самого хазяїна. Коли імунна система реагує на дуже низькі рівні антигену, на які вона зазвичай не повинна реагувати, виникає реакція гіперчутливості. Вважається, що гіперчутливість є причиною алергії та деяких аутоімунних захворювань.
Реакції гіперчутливості можна розділити на чотири типи:
- Гіперчутливість 1 типу включає поширені імунні розлади, такі як астма, алергічний риніт (сінна лихоманка), екзема, кропив'янка та анафілаксія. В усіх цих реакціях беруть участь антитіла IgE, які потребують реакції Th2 під час розвитку Т-хелперів. Профілактичні методи лікування, такі як кортикостероїди та монтелукаст, зосереджені на пригніченні тучних або інших алергічних клітин. Т-клітини не відіграють головної ролі під час фактичної запальної реакції. Важливо відзначити, що числовий розподіл «типів» гіперчутливості не корелює (і абсолютно не пов'язаний) з «реакціями» в моделі Th.
- Гіперчутливість 2 і 3 типу супроводжуються ускладненнями від аутоімунних або низькоафінних антитіл. В обох цих реакціях Т-клітини можуть грати роль співучасника у генерації цих аутоспецифічних антитіл, хоча деякі з цих реакцій при гіперчутливості 2 типу можна вважати нормальними для здорової імунної системи (наприклад, реакції на резус-фактор під час пологів є нормальною імунною відповіддю проти антигенів плода). Розуміння ролі Т-хелперів у цих реакціях обмежене, але зазвичай вважають, що цитокіни Th2 сприятимуть таким розладам. Наприклад, дослідження показали, що вовчак (СЧВ) та інші аутоімунні захворювання подібної природи можуть бути пов'язані з виробленням цитокінів Th2.
- Гіперчутливість 4 типу, також відома як гіперчутливість уповільненого типу, викликається надмірною стимуляцією імунних клітин, як правило, лімфоцитів і макрофагів, що призводить до хронічного запалення та вивільнення цитокінів. Антитіла не грають безпосередньої ролі в цьому типі алергії. Т-клітини відіграють важливу роль у цій гіперчутливості, оскільки вони активуються проти самого подразника та сприяють активації інших клітин; зокрема макрофаги через цитокіни Th1.
Інші види клітинної гіперчутливості включають аутоімунні захворювання, опосередковані цитотоксичними Т-клітинами, та подібні явище — відторгнення трансплантата. Т-хелпери необхідні для розвитку цих захворювань. Щоб створити достатню кількість аутореактивних цитотоксичних Т-клітин, має вироблятися інтерлейкін-2, який постачається Т-хелперами. Т-хелпери за допомогою цитокинів (таких як гамма-інтерферон) також можуть стимулювати клітини, такі як природні кілери та макрофаги, заохочуючи їх, за певних обставин, вбивати клітини господаря.
Механізм, який використовують цитотоксичні Т-клітини під час аутоімунітету, майже ідентичний їх реакції проти вірусів, а деякі віруси вважаються причиною аутоімунних захворювань, таких як цукровий діабет 1 типу. Таке аутоімунне захворювання виникає через те, що системи розпізнавання антигену хазяїна виходять з ладу, й імунна система помилково вважає, що антиген господаря є чужорідним. В результаті цитотоксичні Т-клітини розглядають власну клітину, що представляє цей антиген, як інфіковану, і намагаються знищувати всі власні клітини (або, у разі відторгнення трансплантата, орган для трансплантації), які експресують цей антиген.
Частина цього розділу є спрощенням. Багато аутоімунних захворювань більш складні. Добре відомим прикладом є ревматоїдний артрит, при якому відомо, що і антитіла, й імунні клітини відіграють роль у пошкодженні. Як правило, імунологія більшості аутоімунних захворювань недостатньо вивчена.
ВІЛ-інфекція
Мабуть, найкращим прикладом важливості Т-хелперів є інфекція вірусу імунодефіциту людини (ВІЛ). ВІЛ в основному спрямований проти лімфоїдних CD4+ Т-клітин, але може інфікувати й інші клітини, які експресують CD4, такі як макрофаги та дендритні клітини (обидві групи експресують CD4 на низьких рівнях).
Було припущено, що під час безсимптомної фази ВІЛ-інфекції вірус має відносно низьку спорідненість до Т-клітин (і має більш високу спорідненість до макрофагів), що призводить до повільного знищення CD4+ Т-клітин власною імунною системою. Спочатку їх втрата компенсується за рахунок продукції нових Т-хелперів в тимусі. Однак, як тільки вірус стає лімфотропним (або Т-тропним), він починає інфікувати CD4+ Т-клітини набагато ефективніше (ймовірно, через зміну корецепторів, з якими він зв'язується під час інфекції), і імунна система перевантажується. Слід зазначити, що недавні дослідження показують, що лише ~5 % інфікованих CD4+ Т-клітин лімфоїдного походження, на які спрямований ВІЛ, активно продукують сам вірус. Більше 95 % заражених CD4+ Т-клітин, перебувають у стані спокою і не можуть підтримувати продуктивну інфекцію. Ці клітини піддаються абортивному зараженню ВІЛ.[38] Їх загибель ініціюються, коли вона виробляє проміжну чужорідну ДНК ВІЛ та сама ініціює «суїцид» в спробі захистити організм. У ній в інфламасомі активуєтьсякаспаза-1, тим самим викликається піроптоз (надзапальна форма запрограмованої смерті клітин).[39][40][41]
У цей момент виникає хронічне запалення, і рівні функціональних Т-хелперів починають знижуватися, врешті-решт до точки, коли їх популяція занадто мала, щоб розпізнати весь спектр антигенів, які потенційно можуть бути виявлені. Виснаження Т-хелперів і розвиток хронічного запалення є характерними процесами в патогенезі ВІЛ, які сприяють прогресуванню до синдрому набутого імунного дефіциту (СНІД). Зниження кількості Т-хелперів до рівня менше 200 клітин/мкл у крові під час СНІДу, дозволяє різним патогенам уникнути розпізнавання, що дозволяє опортуністичним інфекціям, на які зазвичай реагують Т-хелпери, обійти імунну систему.[42] Опортуністичні інфекції взагалі виникають тому, що відповідь Т-хелперів абсолютно необхідна для їх усунення. Тяжкість і тривалість більшості інших інфекцій збільшується, оскільки дефіціт Т-хелперів обумовлює менш ефективну імунну відповідь.
Через залежність від Т-хелперів особливо страждають від СНІДу два компоненти імунної системи:
- Цитотоксичні Т-клітини не отримують належної стимуляції, що робить хворих на СНІД дуже сприйнятливими до більшості вірусів, включаючи сам ВІЛ. Це знижує знищення заражених CD4+ Т-клітин і призводить до того, що вірус виробляється протягом більш тривалого періоду, збільшуючи проліферацію вірусу та прискорюючи розвиток захворювання.
- Значно зменшується процес перемикання класу антитіл. Імунна система втрачає здатність покращувати спорідненість своїх антитіл і не може генерувати В-клітини, які можуть виробляти групи антитіл, такі як IgG та IgA. Ці ефекти насамперед пов'язані з втратою будь-якого Т-хелпера, який може правильно взаємодіяти з В-лімфоцитом. Іншим симптомом СНІДу є зниження рівня антитіл через зниження продукції цитокінів Th2 (і менших взаємодій Т-хелперів). Усі ці ускладнення призводять до підвищеної вразливості до агресивних бактеріальних інфекцій, особливо в ділянках тіла, недоступних для антитіл IgM.
Якщо пацієнт не реагує (або не отримує) на лікування ВІЛ, він зазвичай вмирає або від раку, або від інфекцій. Імунна система, нарешті, досягає точки, коли вона більше не координується або не стимулюється для боротьби з хворобою.
Інгібування розмноження Т-хелперів під час ВІЛ-інфекції може відбуватися через мікробну транслокацію IL10-залежним шляхом. Активація PD-1, який експресується на активованих моноцитах за допомогою його ліганду PD-L1, індукує продукцію IL-10, який пригнічує функцію Т-хелперів.[43]
COVID-19
При коронавірусній хворобі 2019 (COVID-19) кількість В-клітин, природних кілерів і усіх лімфоцитів знижується, але CD4+, і CD8+ клітини зменшуються набагато сильніше.[44] Низький рівень CD4+ передбачав більшу ймовірність госпіталізації у відділення інтенсивної терапії, а кількість CD4+-клітин була єдиним параметром, який передбачав тривалість кліренсу (виведення з організму) вірусної РНК.[44] Незважаючи на знижений рівень CD4+-клітин, пацієнти з тяжким перебігом COVID-19 мали вищі рівні Th1 CD4 +-клітин, ніж пацієнти з помірним перебігом захворюванням.[45]
Див. також
- Співвідношення CD4+/CD8+
- CD4+ Т-клітини та протипухлинний імунітет
- CD8+ Т-клітини
- Вакцина проти раку, спрямована на CD4+ Т-клітини
Посилання
- Foundational Model of Anatomy
- Identification of a novel surface protein on activated CD4+ T cells that induces contact-dependent B cell differentiation (help). The Journal of Experimental Medicine 175 (4): 1091–101. April 1992. PMC 2119166. PMID 1348081. doi:10.1084/jem.175.4.1091. Проігноровано невідомий параметр
|vauthors=(довідка) - Molecular interactions mediating T-B lymphocyte collaboration in human lymphoid follicles. Roles of T cell-B-cell-activating molecule (5c8 antigen) and CD40 in contact-dependent help. Journal of Immunology 149 (12): 3817–26. December 1992. PMID 1281189. Проігноровано невідомий параметр
|vauthors=(довідка) - T-BAM/CD40-L on helper T lymphocytes augments lymphokine-induced B cell Ig isotype switch recombination and rescues B cells from programmed cell death. Journal of Immunology 152 (5): 2163–71. March 1994. PMID 7907632. Проігноровано невідомий параметр
|vauthors=(довідка) - Entrez Gene: CD40LG CD40 ligand (TNF superfamily, member 5, hyper-IgM syndrome).
- Sullivan, Kathleen E. (Professor of pediatrics) Stiehm, E. Richard, 1933- (2020). Stiehm's immune deficiencies inborn errors of immunity. Academic Press. ISBN 978-0-12-817295-7. OCLC 1156337055.
- Oestreich, Kenneth J.; Weinmann, Amy S. (12 жовтня 2012). Master regulators or lineage-specifying? Changing views on CD4+ T cell transcription factors. Nature Reviews Immunology 12 (11): 799–804. ISSN 1474-1733. PMC 3584691. PMID 23059426. doi:10.1038/nri3321.
- Ma, Cindy S. Chew, Gary Y.J. Simpson, Nicholas Priyadarshi, Archana Wong, Melanie Grimbacher, Bodo Fulcher, David A. Tangye, Stuart G. Cook, Matthew C. (2008). Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. The Journal of Experimental Medicine (The Rockefeller University Press) 205 (7): 1551–1557. OCLC 679066657. PMC 2442632. PMID 18591410. doi:10.1084/jem.20080218.
- Ma, Cindy S.; Avery, Danielle T.; Chan, Anna; Batten, Marcel; Bustamante, Jacinta; Boisson-Dupuis, Stephanie; Arkwright, Peter D.; Kreins, Alexandra Y. та ін. (26 квітня 2012). Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood 119 (17): 3997–4008. ISSN 0006-4971. PMC 3355712. PMID 22403255. doi:10.1182/blood-2011-11-392985. Проігноровано невідомий параметр
|doi-access=(довідка); - Fink, Pamela J. (21 березня 2013). The Biology of Recent Thymic Emigrants. Annual Review of Immunology 31 (1): 31–50. ISSN 0732-0582. PMID 23121398. doi:10.1146/annurev-immunol-032712-100010.
- The full spectrum of human naive T cells. Nature Reviews. Immunology 18 (6): 363–373. March 2018. PMID 29520044. doi:10.1038/s41577-018-0001-y. Проігноровано невідомий параметр
|vauthors=(довідка) - Neonatal thymectomy reveals differentiation and plasticity within human naive T cells. The Journal of Clinical Investigation 126 (3): 1126–36. March 2016. PMC 4767338. PMID 26901814. doi:10.1172/JCI84997. Проігноровано невідомий параметр
|vauthors=(довідка) - Roche, Paul A.; Furuta, Kazuyuki (27 лютого 2015). The ins and outs of MHC class II-mediated antigen processing and presentation. Nature Reviews Immunology 15 (4): 203–216. ISSN 1474-1733. PMC 6314495. PMID 25720354. doi:10.1038/nri3818.
- Unanue, Emil R.; Turk, Vito; Neefjes, Jacques (11 травня 2016). Variations in MHC Class II Antigen Processing and Presentation in Health and Disease. Annual Review of Immunology 34 (1): 265–297. ISSN 0732-0582. PMID 26907214. doi:10.1146/annurev-immunol-041015-055420.
- Wieczorek, Marek; Abualrous, Esam T.; Sticht, Jana; Álvaro-Benito, Miguel; Stolzenberg, Sebastian; Noé, Frank; Freund, Christian (17 березня 2017). Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Frontiers in Immunology 8: 292. ISSN 1664-3224. PMC 5355494. PMID 28367149. doi:10.3389/fimmu.2017.00292. Проігноровано невідомий параметр
|doi-access=(довідка) - Trolle, Thomas; McMurtrey, Curtis P.; Sidney, John; Bardet, Wilfried; Osborn, Sean C.; Kaever, Thomas; Sette, Alessandro; Hildebrand, William H. та ін. (15 лютого 2016). The Length Distribution of Class I–Restricted T Cell Epitopes Is Determined by Both Peptide Supply and MHC Allele–Specific Binding Preference. The Journal of Immunology (англ.) 196 (4): 1480–1487. ISSN 0022-1767. PMC 4744552. PMID 26783342. doi:10.4049/jimmunol.1501721.
- Murphy, Kenneth. (2017). Janeway's immunobiology. Garland Science. ISBN 978-0-8153-4551-0. OCLC 1020120603.
- Guy, Bruno (July 2007). The perfect mix: recent progress in adjuvant research. Nature Reviews Microbiology (англ.) 5 (7): 396–397. ISSN 1740-1534. PMID 17558426. doi:10.1038/nrmicro1681.
- Hammer, Gianna Elena; Ma, Averil (21 березня 2013). Molecular Control of Steady-State Dendritic Cell Maturation and Immune Homeostasis. Annual Review of Immunology 31 (1): 743–791. ISSN 0732-0582. PMC 4091962. PMID 23330953. doi:10.1146/annurev-immunol-020711-074929.
- Zamoyska, Rose (September 2007). Why Is There so Much CD45 on T Cells?. Immunity 27 (3): 421–423. ISSN 1074-7613. PMID 17892852. doi:10.1016/j.immuni.2007.08.009. Проігноровано невідомий параметр
|doi-access=(довідка) - Courtney, Adam H.; Shvets, Alexey A.; Lu, Wen; Griffante, Gloria; Mollenauer, Marianne; Horkova, Veronika; Lo, Wan-Lin; Yu, Steven та ін. (22 жовтня 2019). CD45 functions as a signaling gatekeeper in T cells. Science Signaling (англ.) 12 (604): eaaw8151. ISSN 1945-0877. PMC 6948007. PMID 31641081. doi:10.1126/scisignal.aaw8151.
- Molecular and biological characterization of a murine ligand for CD40. Nature 357 (6373): 80–2. May 1992. Bibcode:1992Natur.357...80A. PMID 1374165. doi:10.1038/357080a0. Проігноровано невідомий параметр
|vauthors=(довідка) - A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci U S A 89 (14): 6550–4. July 1992. Bibcode:1992PNAS...89.6550N. PMC 49539. PMID 1378631. doi:10.1073/pnas.89.14.6550. Проігноровано невідомий параметр
|vauthors=(довідка); Проігноровано невідомий параметр|doi-access=(довідка) - Thymic and Postthymic Regulation of Naïve CD4(+) T-Cell Lineage Fates in Humans and Mice Models. Mediators of Inflammation 2016: 9523628. 2016. PMC 4904118. PMID 27313405. doi:10.1155/2016/9523628. Проігноровано невідомий параметр
|vauthors=(довідка); Проігноровано невідомий параметр|doi-access=(довідка) - CD4 T cells: fates, functions, and faults. Blood 112 (5): 1557–69. September 2008. PMC 2518872. PMID 18725574. doi:10.1182/blood-2008-05-078154. Проігноровано невідомий параметр
|vauthors=(довідка) - GATA3: a master of many trades in immune regulation. Trends in Immunology 35 (6): 233–42. June 2014. PMC 4045638. PMID 24786134. doi:10.1016/j.it.2014.04.002. Проігноровано невідомий параметр
|vauthors=(довідка) - Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: a critical review. Journal of Autoimmunity 57 (6): 1–13. February 2015. PMC 4340844. PMID 25578468. doi:10.1016/j.jaut.2014.12.002. Проігноровано невідомий параметр
|vauthors=(довідка) - Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol 8 (8): 825–34. 2007. PMID 17589510. doi:10.1038/ni1482. Проігноровано невідомий параметр
|vauthors=(довідка) - Pharmacology. Edinburgh: Churchill Livingstone. 2003. ISBN 978-0-443-07145-4. Page 223
- Interleukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity 31 (2): 209–19. August 2009. PMC 2791889. PMID 19646904. doi:10.1016/j.immuni.2009.05.012. Проігноровано невідомий параметр
|vauthors=(довідка) - Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nature Immunology 6 (11): 1123–32. November 2005. PMID 16200070. doi:10.1038/ni1254. Проігноровано невідомий параметр
|vauthors=(довідка) - Fate mapping of IL-17-producing T cells in inflammatory responses. Nature Immunology 12 (3): 255–63. March 2011. PMC 3040235. PMID 21278737. doi:10.1038/ni.1993. Проігноровано невідомий параметр
|vauthors=(довідка) - Multiparameter grouping delineates heterogeneous populations of human IL-17 and/or IL-22 T-cell producers that share antigen specificities with other T-cell subsets. European Journal of Immunology (UPMC Paris 06 Institut National de la Santé et de la Recherche Médicale (Inserm) UMR-S 945) 41 (9): 2596–605. September 2011. PMID 21688259. doi:10.1002/eji.201041131. Проігноровано невідомий параметр
|vauthors=(довідка); Проігноровано невідомий параметр|doi-access=(довідка) - Helper T cell diversity and plasticity. Current Opinion in Immunology 24 (3): 297–302. June 2012. PMC 3383341. PMID 22341735. doi:10.1016/j.coi.2012.01.014. Проігноровано невідомий параметр
|vauthors=(довідка) - (Veldhoen and Stockinger, Dardalhon and Kuchroo both papers from Nature Immunology 2008)
- Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401 (6754): 708–712. 1999. Bibcode:1999Natur.401..708S. PMID 10537110. doi:10.1038/44385. Проігноровано невідомий параметр
|vauthors=(довідка) - CD4+ virtual memory: Antigen-inexperienced T cells reside in the naïve, regulatory, and memory T cell compartments at similar frequencies, implications for autoimmunity. Journal of Autoimmunity 77 (2): 76–88. 2017. PMC 6066671. PMID 27894837. doi:10.1016/j.jaut.2016.11.001. Проігноровано невідомий параметр
|vauthors=(довідка) - Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143 (5): 789–801. November 2010. PMC 3026834. PMID 21111238. doi:10.1016/j.cell.2010.11.001. Проігноровано невідомий параметр
|vauthors=(довідка) - Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505 (7484): 509–14. January 2014. Bibcode:2014Natur.505..509D. PMC 4047036. PMID 24356306. doi:10.1038/nature12940. Проігноровано невідомий параметр
|vauthors=(довідка) - IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343 (6169): 428–32. January 2014. Bibcode:2014Sci...343..428M. PMC 3976200. PMID 24356113. doi:10.1126/science.1243640. Проігноровано невідомий параметр
|vauthors=(довідка) - Zhang, Chao; Song, Jin-Wen; Huang, Hui-Huang; Fan, Xing; Huang, Lei; Deng, Jian-Ning; Tu, Bo; Wang, Kun та ін. (15 березня 2021). NLRP3 inflammasome induces CD4+ T cell loss in chronically HIV-1–infected patients. The Journal of Clinical Investigation (англ.) 131 (6). ISSN 0021-9738. PMC 7954596. PMID 33720048. doi:10.1172/JCI138861. Проігноровано невідомий параметр
|doi-access=(довідка); - CD4 Count. www.aids.gov. Процитовано 30 квітня 2015.
- Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nature Medicine 16 (4): 452–9. April 2010. PMC 4229134. PMID 20208540. doi:10.1038/nm.2106. Проігноровано невідомий параметр
|vauthors=(довідка) - Lymphocyte Subset Counts in COVID-19 Patients: A Meta-Analysis. Cytometry Part A 97 (8): 772–776. 2020. PMC 7323417. PMID 32542842. doi:10.1002/cyto.a.24172. Проігноровано невідомий параметр
|vauthors=(довідка) - Perlman S (2020). COVID-19 poses a riddle for the immune system. Nature 584 (782): 345–346. PMID 32807916. doi:10.1038/d41586-020-02379-1. Проігноровано невідомий параметр
|doi-access=(довідка)
